原料药生产分为哪几个阶段,各有什么特点-原料药生产的一般特点有哪些

药品生产过程质量保证体系:
(一)人员生产过程质量保证体系控制系统。
(二)公用工程控制系统。
(三)备控制系统。
(四)物料控制系统。
(五)生产过程控制系统。
(六)质量检测控制系统。
(七)文件控制系统。
(八)验证控制系统。
(九)用户抱怨控制系统。
药品生产是指将原料加工制成能够供医疗使用的药品的过程。药品生产的过程通常可分为原料药生产阶段和将原料药制成一定剂型的制剂生产阶段。此外,对某些药物来说,还包括制药中间体的生产。
药品的性质、作用及其特点客观上决定了现代的药品生产具有以下特点:
1.机械化、自动化程度要求高。对药品而言,生产人员本身就是污染源。因此,药品生产要求较高的机械化、自动化程度。
2.生产过程中使用仪器设备较多,且生产设备具有较强的多用性(即用于生产多种药品)。
3卫生要求严格生产车国的卫生洁争程度及厂区的卫生状况都会对药品质量产生较大影响,甚至不同品种或一品种不同批次的品之间都为污染源,因此,药品生产对生产环境的卫生要求十分严格,厂区、路面及运输等不得对药品的生产造成污染,生产人员、设备及药品的包装物等均不得对药品造成污染。
4.对生产条件有较高的要求。温度、湿度、空气洁净度等直接影响药品质量,是药品生产过程中需要严格控制的因素。
5.产品有严格的质量基线要求,药品不允许有 等外品”、"处理品”等,必须是符合药品标的合格品:目产品一出现质量问题,通常不能饭修”。因此,客上要求药品生产外于零美错率状态。
6.产品的生产及其内在质量检验的专业业性较强。
7.生产环节多、生产过程较为复杂。
8.通常产生较多的“三废”。
9.固定成本较高、规模的经济性较强。
10.产品的种类、规格、剂型繁多。
软膏剂的制备方法有哪些?各有何特点?如何选用
制药业独特的双重角色是原料药和成品药。
原料药是生产成品药的主要原材料,不能直接给患者使用,成品药就是用原料药生产的可以直接给患者使用的药。
化学药品的生产部门,由原料药生产和药物制剂生产两部分组成。原料药是药品生产的物质基础,但必须加工制成适合于服用的药物制剂,才成为药品。在中国,制药工业还包括传统中药的生产。制药工业既是国民经济的一个部门,又是一项治病、防病、保健、计划生育的社会福利事业。
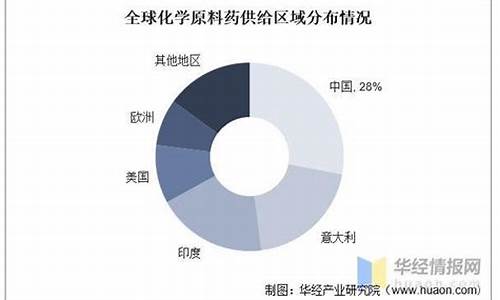
原料药品种众多,其生产方法各不相同,有全合成法,有发酵法兼用提炼技术,有合成法兼用生物技术,有发酵产品再进行化学加工,也有主要采用分离提纯方法。
原料药生产的一般特点:
1、生产流程长、工艺复杂。
2、每一产品所需的原辅材料种类多,许多原料和生产过程中的中间体是易燃、易爆、有毒或腐蚀性很强的物质,对防火、防爆、劳动保护以及工艺和设备等方面有严格的要求。
3、产品质量标准高(纯度高、杂质可允许的含量极微),对原料和中间体要严格控制其质量。
4、物料净收率很低,往往几吨至上百吨的原料才生产一吨产品,因而副产品多,三废也多。
5、药物品种多、更新快、新药开发工作的要求高、难度大、代价高、周期长。制剂生产则需要有适合条件的人员、厂房、设备、检验仪器和良好的卫生环境以及各种必需的制剂辅料和适用的内、外包装材料相配合。
医药中间体是什么意思
制备方法:
1.研磨法
适用于油脂性基质的软膏剂的制备。把半固体状态的油脂性基质和研细过筛过的药物粉末直接研磨混合制备软膏剂的方法。制备时将药物研细过筛后,先用等量基质研匀,然后等量递加其余基质至全量,研匀即得。本法适用于少量软膏剂的制备,而且药物不溶于基质中的情况。在实验室制备时可在乳钵中研磨;大量生产时可用电动研钵制备。
2.乳化法
适合于乳剂型软膏剂的制备。将处方中的油脂性和油溶性成分一起加热至80℃:左右成油溶液;另将水溶性成分溶于水中,并加热至80℃:左右成水溶液。两相混合时为了防止油相中的固体成分过早析出或凝结,使水相温度略高于油相温度。将水相逐渐加入油相中,边加边搅拌,直至冷凝。大量生产时,在温度降低至30℃时再通过胶体磨或软膏研磨机使更细腻均匀。
乳化法中水、油两相的混合有三种方法:①两相同时掺合,适用于大批量的机械操作;②分散相加到连续相中,适用于含小体积分散相的乳剂系统;③连续相加到分散相中,适用于多数乳剂系统。如制备O/W型乳膏基质时,在搅拌下将水相缓缓加到油相内,开始水相的量小于油相,先形成W/O型乳液,继续把水相加人油相时,乳液蒙古度继续增加,直到W/O型乳液水相的体积增加到最大限度,超过此限,乳液蒙古度降低,发生转型而成为O/W型乳液,-使内相(油相)分散的更细,冷却后形成O/W型乳剂型基质。
乳化法使用的乳化机有三种类型:乳化搅拌机、胶体磨和均质机。
影响乳剂型基质质量的因素:①设备搅拌速度的影响,如搅拌速度过小,达不到充分海合的目的,搅拌速度过大,会将气泡带入体系,使之成为三相体系,使乳状液不稳定;②乳化温度的影响,乳化温度取决于两相中所含有高熔点物质的熔点,另一方面在乳化过程中基质的黏度会增加很多,提高温度,降低勃度有利于基质各成分的混合均匀,一般控制在75-85℃之间,如有转相泪度,则乳化温度应控制在转相温度附近;③乳化时间的影响,要根据油相与水相的容积比,两相的蒙古度及生成乳状液的黏度,乳化剂的类型及用量,乳化温度来确定。实际工作中乳化时间与乳化设备的效率紧密相连,如用均质机(3000r/min)进行乳化,仅需用3-10 min。
3.熔融法
适用于油脂性基质的制备,在基质处方中含有熔点高的组分时,在常温下不能混合均匀。熔融法,先将熔点较高的基质,如蜂蜡(62-67℃)、石蜡(48-58℃)、硬脂酸(55-60℃)等熔化,再按熔点高低依次加入熔化、搅拌混合均匀,直至冷凝。制备的软膏如果不够细腻,需要通过研磨机进一步研匀,使之无颗粒的沙砾感。
以下为2015版药典第四部0109对软膏剂的要求:
软膏剂 系指原料药物与油脂性或水溶性基质混合制成的均匀的半固体外用制剂。
因原料药物在基质中分散状态不同,分为溶液型软膏剂和混悬型软膏剂。溶液型软膏剂为原料药物溶解(或共熔)于基质或基质组分中制成的软膏剂;混悬型软膏剂为原料药物细粉均匀分散于基质中制成的软膏剂。
乳膏剂 系指原料药物溶解或分散于乳状液型基质中形成的均匀半固体制剂。
乳膏剂由于基质不同,可分为水包油型乳膏剂和油包水型乳膏剂。
软膏剂、乳膏剂在生产与贮藏期间应符合下列有关规定。
一、乳膏剂、乳膏剂选用基质应根据各剂型特点、原料药物的性质、制剂的疗效和产品的稳定性。基质也可由不同类型基质混合组成。
软膏剂基质可分为油脂性基质和水溶性基质。油脂性基质常用的有凡士林、石蜡、液状石蜡、硅油、蜂蜡、硬脂酸、羊毛脂等;水溶性基质主要有聚乙二醇。乳膏剂常用的乳化剂可分为水包油型和油包水型。水包油型乳化剂有钠皂、三乙醇胺皂类、脂肪醇硫酸(酯)钠类和聚山梨酯类;油包水型乳化剂有钙皂、羊毛脂、单甘油酯、脂肪醇等。
二、软膏剂、乳膏剂基质应均匀、细腻,涂于皮肤或黏膜上应无刺激性。软膏剂中不溶性原料药物,应预先用适宜的方法制成细粉,确保粒度符合规定。
三、软膏剂、乳膏剂根据需要可加入保湿剂、抑菌剂、增稠剂、稀释剂、抗氧剂及透皮促进剂。除另有规定外,加入抑菌剂的软膏剂、乳膏剂在制剂确定处方时,该处方的抑菌效力应符合抑菌效力检查法(通则1121)的规定。
四、软膏剂、乳膏剂应具有适当的黏稠度,应易涂布于皮肤或黏膜上,不融化,黏稠度随季节变化应很小。
五、软膏剂、乳膏剂应无酸败、异臭、变色、变硬等变质现象。乳膏剂不得有油水分离及胀气现象。
六、除另有规定外,软膏剂应避光密封贮存。乳膏剂应避光密封置25°C以下贮存,不得冷冻。
七、软膏剂、乳膏剂所用内包装材料,不应与原料药物或基质发生物理化学反应,无菌产品的内包装材料应无菌。软膏剂、乳膏剂用于烧伤治疗如为非无菌制剂的,应在标签上标明“非无菌制剂”;产品说明书中应注明“本品为非无菌制剂”,同时在适应证下应明确“用于程度较轻的烧伤(Ⅰ°或浅Ⅱ°)”;注意事项下规定“应遵医嘱使用”。
除另有规定外,软膏剂、乳膏剂应进行以下相应检查。
粒度除另有规定外,混悬型软膏剂、含饮片细粉的软膏剂照下述方法检査,应符合规定。
检査法取供试品适量,置于载玻片上涂成薄层,薄层面积相当于盖玻片面积,共涂3片,照粒度和粒度分布测定法(通则0982第一法)测定,均不得检出大于18CVm的粒子。
装量照最低装量检査法(通则0942)检查,应符合规定。
无菌用于烧伤[除程度较轻的烧伤(Ⅰ°或浅Ⅱ°外)]或严重创伤的软膏剂与乳膏剂,照无菌检查法(通则1101)检查,应符合规定。
微生物限度除另有规定外,照非无菌产品微生物限度检查:微生物计数法(通则1105)和控制菌检查法(通则1106)及非无菌药品微生物限度标准(通则1107)检查,应符合规定。
口服原料药,注射用原料药,一般原料药,3者的区别和定义
摘要:医药中间体指的是用于药品合成工艺过程中的一些化工原料或化工产品,为药品合成的原料,非药品,要求比药品低。在生产中,医药中间体的生产特点有经营灵活,投资规模不大、产品更新速度快、生产企业地域分布比较集中等。具体的医药中间体是什么意思以及医药中间体的生产特点有哪些,咱们继续往下看看吧!一、医药中间体是什么意思
所谓医药中间体,实际上是一些用于药品合成工艺过程中的一些化工原料或化工产品。
这种化工产品,不需要药品的生产许可证,在普通的化工厂即可生产,只要达到一些的级别,即可用于药品的合成。
其实药品的合成亦属于化工类,只不过比一般的化工厂要求要严格,所以药厂需要GMP认证,而医药中间体为药品合成的原料,非药品,所以相对来说要求比药品低,生产厂不需要药品GMP认证。
二、医药中间体的生产特点有哪些
纵观整个行业,目前我国医药中间体生产有六大特点:
1、生产企业多为私营企业,经营灵活,投资规模不大,基本上在数百万到一两千万元之间。
2、生产企业地域分布比较集中,主要分布在以浙江台州和江苏金坛为中心的地区。
3、随着国家对环保问题的日益重视,生产企业建设环保处理设施的压力增大。
4、产品更新速度快。一个产品一般面市3~5年后,其利润率便大幅度下降,这迫使企业必须不断开发新产品或不断改进生产工艺,才能保持较高的生产利润。
5、由于医药中间体的生产利润高于化工产品,两者的生产过程又基本相同,于是便有越来越多的小型化工企业加入了生产医药中间体行列,导致行业内无序竞争日益激烈。
6、与原料药相比,生产中间体利润率偏低,而原料药与医药中间体的生产过程又相似,因此,部分企业已不仅仅生产中间体,还利用自身优势,开始生产原料药。
制药工程技术的特点
注射用原料药:需要在万级车间生产
口服原料药:需要在10万级车间生产
一般原料药:需要在30万级车间生产
这是根据药品企业洁净室(区)的特点,针对不同情况制订的标准。
医药原料药申报中易出现的几点问题
制药工程的特点 制药工业 主要指化学药品的生产部门,由原料药生产和药物制剂生产两部分组成。原料药是药品生产的物质基础, 但必须加工制成适合于服用的药物制剂才成为药品。
一、高度的科学性、技术性。二、分工细致明确、质量标准规范。
三、生产过程复杂、品种繁多四、生产过程的连续性五、高投入、高产出
制药工程专业毕业后可到制药工程(或医药生物技术)领域相关的生产企业、营销企业、科研院所、药品监督管理部门等企、事业单位从事药品生产、管理、营销、检验监督和研发等工作。也适于报考生物技术、药学及相关专业的研究生。
原料药的质量是药品质量的基础,其质量不能仅依靠最终的质量标准来控制和保证,还必须对整个制备过程加以控制。结合目前原料药的审报情况,分析整理出以下问题,提请申报者关注:(一)合成工艺 1、缺乏对合成用起始原料、关键原料的合理控制。起始原料内控标准的制订不仅仅是研究资料完整性的一个方面,更重要的是有利于申报单位加强对原料药合成的 起点控制,最大限度地降低可能引入的杂质,保证终产品的纯度。审评中发现申报资料部分研究单位往往忽视制订起始原料的内控标准,或者即使制定,也不是结合 起始原料的工艺设定合理的质控项目(比如未结合工艺,制定相应的杂质控制以及残留溶剂控制)。2、缺少反应终点的监测方法与中间体质控方法。对于反应终点的监测以及中间体的控制,是构成产品质量控制体系的一部分重要内容。建议研发者尽量采用TLC等方法监测反应进程,对关键中间体应建立HPLC法等定量分析方法进行质控,以保证工艺与质量的稳。尤其需要强调的是目前手性原料的合成中,对引入手性的原料、中间体的控制过于粗略。研究单位多采用比旋度的方法对手性原料药合成中的关键原料及中间体进行 光学活性控制,但比旋度对光学纯度的质控而言,是个较为粗略的指标,其数值受样品的化学纯度、水分等的影响而会产生较大的波动,无法较准确体现样品的光学 纯度。同时,由于手物尤其是多个手性中心的药物空间结构确证以及质量控制仅依靠终点控制有一定难度,通常尚需结合起始材料以及中间体的情况进行判断, 故建议此类药物合成中采用手性HPLC法、毛细管电泳等更具专属性的方法控制相关样品的光学纯度。(二)、结构确证 1、结构确证中的对照品问题:结构确证不一定都要使用对照品,在没有对照品时,只需根据结构确证的一般原则:在全面分析化合物结构特征的基础上,结合制备 工艺、文献数据等已有的研究信息,选择针对性强的分析方法来确证化合物的结构。如果选用对照品,则需关注对照品选择的合理性:如以自研产品精制后样品作为 对照品,对结构确证而言,无专属性及特异性意义,因此并不适宜作为结构确证时的对照品;以上市制剂中提取、精制的原料为对照品,可作为部分结构确证时使 用,但由于提取、精制用溶剂及方法的差异,此类对照品与样品有可能存在晶型等方面的差异,故不适宜作为DSC、TG、粉末X-射线衍射等测定时的对照品。2、对口服固体制剂所用的难溶性原料药,缺少对样品晶型的研究晶型不同,可能会影响到产品的稳定性以及溶解性(最终影响生物利用度),为减少临床研究 的风险,建议对难溶物加强晶型研究。可采用不同的精制方法获取具有潜在晶型差异的样品,并对这些样品进行IR或粉末X-射线衍射测定以确定样品是否具 有多晶型,对具有多晶型的样品尽可能选择成熟路线制备晶型热力学稳定的样品作为制剂的原料,同时应兼顾不同晶型样品的溶解度。(三)质量研究与质量标准 该部分研究是存在问题最多的部分,最突出的是有关物质、残留溶剂方法建立的合理性、可操作性及质量可控性。1、有关物质检查:有关物质检查,包括对产品中残留合成原料、中间体、副产物及可能的降解产物的检查,是控制药品质量的重要指标,同时也是药品稳定性评价中需重点考察的项目。其方法学研究需关注以下几个项目:(1)有关物质检查波长的选择:当采用HPLC法,检测器为紫外检测器时,检测波长选择是否合理直接影响到杂质种类、数量的检出,因此检测波长的选择是 方法学研究的重要内容。审评中常见的问题包括: 直接或间接地以主成分的最大吸收波长作为检测波长,由于有关物质检查的对象是杂质,若将主药的最大吸收波长确定为检测波长,则杂质在此波长下的吸收可能偏 低,某些杂质甚至无吸收,这样会造成对杂质含量的低估甚至漏检,从而不能反映产品的真实质量。以样品进行破坏性试验(酸、碱、热、光照、氧化等)后的溶液 做紫外扫描,将扫描图谱中最大吸收波长确定为有关物质的检测波长。因破坏性试验后溶液中存在尚未破坏的主药、降解产物、辅料等,此溶液的紫外吸收为各成分 紫外吸收的加和,并不能反映降解产物的紫外吸收特性。由于未破坏主药所占比例较大,故破坏性试验后溶液的最大吸收波长一般仍为主药的最大吸收波长。因此在有关物质检查的波长选择时,首推通过二极管阵列检测器考察合成用原料、各中间体、各降解产物、主药的紫外吸收特征,或至少通过紫外扫描的方法考 证上述各样品的紫外吸收特征,选择杂质与原料相近的响应值处的波长为有关物质检查波长。对于响应值相差较大的杂质,应建立相应的检查方法及检测波长,或采 用加校正因子的自身对照法。(2)流动相筛选及方法学验证:从目前原料药的审报情况来看,在流动相筛选及方法学验证中,采用强力破坏以获得各种降解产物,并考查各降解产物与主药的分 离度的方法已被申报单位接受并认同,易被申报单位忽视的是对合成用原料、中间体与主药间分离度的考证。而对于原料药而言,这恰恰是原料药流动相筛选及考证 的重点,尤其是对于已有国家标准的原料药,验证国家标准终收载的有关物质检查方法是否适合自研产品的检验,由于合成工艺的可能不同,需重点验证合成用起始 原料、中间体同主药的分离情况。2、残留溶剂:原料药中的残留溶剂系指在原料药生产中使用,但在工艺过程中未能完全去除的有机溶剂。申报情况反映对于残留溶剂的检查可能存在以下问题:(1)研究内容不全面:未进行或只进行部分残留溶剂的检测,原料药中残留的有机溶剂特别是二类以上溶剂可能会对药物的安全性产生重大影响,其研究的重要性 不言而喻,由于历史原因,部分国家标准未制定有机溶剂检查项,但随着药检技术的发展和对残留溶剂认识的提高,残留溶剂研究成为必要的研究项目,需根据考察 结果(尤其是大生产样品的检验结果)确定是否应将该项检查定入质量标准。对制剂过程中使用的有机溶剂也建议考察其残留情况,特别是脂质体、缓、控释微丸包 衣过程使用的有机溶剂更应引起注意。一般情况下一类溶剂应禁止使用,如要使用,必须首先进行替代试验,证明在合成工艺中无法用其他溶剂替代。一类不论是在 起先还是后续的反应中使用,均应在产品中检测控制。(2)方法学研究不完整:采用GC顶空法进行测定时,研究过程中往往忽视两个非常重要的参数:顶空的平衡温度和平衡时间。平衡温度影响分配系数,并与平衡 时间相关,略高的平衡温度可以缩短平衡时间。平衡时间本质上取决于被测组分分子从样品基质到气相的扩散速度,由于样品的性质千差万别,因此平衡时间难以预 测,需通过一定平衡温度下的试验确定,以保证样品中有机溶剂的充分释出。通过研究确定合理的平衡温度与平衡时间保是保证顶空法测定残留溶剂结果可靠性的重 要前提。(3)检测器选择不当:气相的检测器用于残留溶剂的测定时最常用的是火焰离子化检测器(FID)、热导检测器(TCD)、电子捕获检测器(ECD)。申报 资料中存在的主要问题是忽视研究对象的结构特征,不加研究及选择地使用某一种检测器,如被研究对象是三氯甲烷、四氯化碳等仅含一个活泼氢或不含活泼氢的化 合物,却采用了FID检测器,导致测定结果的不可靠。(四)稳定性考察1、考察项目设置不合理:稳定性研究的考察项目应选择在药品保存期间易于变化,并可能会影响到药品的质量、安全性和有效性的项目,以便客观、全面地反映药 品的稳定性。根据药品特点和质量控制的要求,尽量选取能灵敏反映药品稳定性的指标。目前申报情况反映,有些申报单位易忽视产品的特点,仅以常规或专属性较 差的考察项目代替样品个性的考察,如对手物不考察光学异构体的变化或仅以比旋度进行粗略考察,对于易吸湿的药物不进行水分或干燥失重检查,无法全面、 真实反映样品的稳定性。2、稳定性研究中采用的方法与质量标准的方法不一致。质量标准中的方法是研究者在研发中,经过比较,认为相对合理的、可控制产品质量的方法,因此,两部分 研究中方法应尽量保持一致,以加强对结果的判断。如产品在研发过程中,质控方法进行了修订,需要分析方法变更前后对检验结果的影响。徐州宏鑫医药化工有限公司位于风景秀丽的徐州工业园区,园区地理位置优越,交通便利。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。