欧盟药品注册有哪些方式-欧盟原料药注册制度

FDA,COS,GOST。
医药一般都要到药监局办理药品方面的进出口许可证回复问题出口到不同国家都需要一些认证的美国FDA食品,药品,保健品,医疗机械,化妆品的监督检验欧盟COS中国主要是针对中草药这样的原料药,欧洲通过该认证,通过欧洲药典来审核该药品是否合法俄罗斯GOST俄罗斯的对药品进口的强制认证,不通过海关都过不了国际通用的GMP国际药品生产通用准则,不过一般国家都根据自己的标准确定标准HACCP美国鉴别,评价和控制对食品安全至关重要的危害的一种体系。
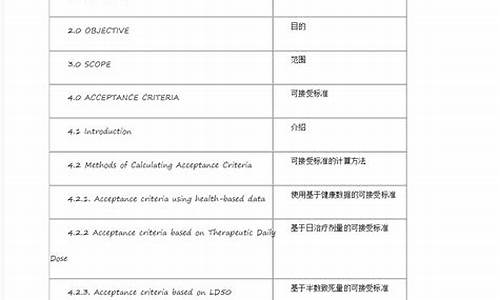
欧盟药品GMP指南的目录
COS
认证和
EDMF
注册的比较
EDMF
和
COS
证书都是原料药进入欧洲市场有效而必需的支持性材料,
二者都是用于证明
制剂产品中所使用的原料药质量的文件以便支持使用该原料药的制剂产品在欧洲的上市申
请(
MAA
);它们之间究竟有什么不同呢?
首先,是评审方式上的不同。
EDMF
是由单个国家的机构评审的,是作为制剂上市许
可申请文件的一部分而与整个制剂的上市许可的申请文件一起进行评审的。针对不同的制
剂,不同的评审机构有不同的侧重,
因而会对文件有不同的要求,
提出不同的问题。无论原
料药物用于哪个制剂的生产,也无论该
EDMF
是否已进行过登记,都要进行重新评审,因
而对我们这些原料药的生产厂家来说是多次申请登记,要花费更多的时间和精力。而
COS
申请文件是由有关当局组成的专家委员会集中评审的,
评审结果将决定是否发给
COS
证书。
一个原料药一旦取得
COS
证书,就可以用于欧洲药典委员会的三十一个成员国内的所有制
剂生产厂家的制剂生产。
其次,针对的情况不同。
EDMF
与使用该原料药的制剂药物的上市许可申请(
MAA
)
不可分离,必须由使用该原料药的欧洲终端用户申请;而
COS
证书则是直接将证书颁发给
原料药的生产厂家,
因此可由原料药生产厂家独立申请
,
并不需要现成的中间商和终端用户,
因而生产厂家在申请过程中更加主动。
第三,适用的范围不同。
EDMF
程序适用于所有的原料药品,只要是原料药,无论是
否已收载入欧洲药典,
都可以通过
EDMF
文件的方式进入欧洲市场,
而
COS
证书只能处理
欧洲药典已收载的物质,
当然不仅是原料药,
也包括生产制剂所用的辅料,
我国的药用辅料
也可以申请
COS
证书。
第四,所要求提供的资料不同。比如
EDMF
文件必须包括药物的稳定性研究资料,而
COS
证书的申请文件并不强求这些资料。
第五,申请的结果不同。申请
COS
证书的结果是直接颁发给原料药的生产厂家一个证
书,
只要将这个证书的复印件提供给欧洲方面的中间商或终端用户,
对方就可以购买我们的
原料药,而
EDMF
文件登记的结果是只告诉制剂生产厂家一个
EDMF
文件的登记号,欧洲
评审机构不会将这个登记号告诉原料药的生产厂家,
原料药的生产厂家只能从负责申请登记
的欧洲药品制剂的生产厂家那儿查询这个登记号。
CoS
(
Certificate
of
Suitability
)指的是欧洲药典适用性认证,目的是考察欧洲药典是否能
够有效地控制进口药品的质量,
这是中国的原料药合法地被欧盟的最终用户使用的另一种注
册方式。
这种注册途径的优点是不依赖于最终用户,
可以由原料药生产厂商独立地提出申请。
中国的原料药生产厂商可以向欧盟药品质量指导委员会(
EDQM
)提交产品的
CoS
认证文
件(
CoS
Dossier
),申请
CoS
证书,同时生产厂商必须要承诺产品生产的质量管理严格
遵循
GMP
标准,
在文件审查和可能的现场考察通过之后,
EDQM
会向原料药品的生产厂商
颁发
CoS
证书。
如果作为最终用户的欧盟成员国制剂生产企业准备采用中国生产的原料时,
只要在注册文件或变更文件中附上该产品的
CoS
证书复印件即可非常容易地获得批准。
欧洲药典适应性认证证书(
CEP
)不仅被所有欧盟成员国所承认,而且被很多承认欧
洲药典地位的国家所认可,如很多欧盟以外的欧洲国家、澳大利亚和中国。
CEP
证书能够
替代
EDMF
文件用于药品上市申请和原料药来源的变更申请。
要求
随着美国、
欧盟和日本三方在药品注册程序和法规上的相互协调,
欧盟在进口的原料药
注册中逐步接近美国
FDA
的偏重现场
GMP
检查的办法,
今后有可能对每一家提出
COS
认
证的生产厂家进行现场的
GMP
检查。
自
1999
年开始,原料药生产企业在申请
COS
认证的技术文件后面必须要附加两封承
诺信,
一封信承诺说产品是按照
GMP
规范进行生产的,
另一封信要承诺同意欧盟的相关审
查机构进行现场检查。如果欧盟
EDQM
的
GMP
审查越来越频繁,甚至最终变成为一种必
要的审查手段,
生产厂家就应当对此做出充分的准备,以使自身的
GMP
管理状况能够适应
欧盟的检查。
欧盟的
GMP
检查与国内的
GMP
认证有以下差别:首先,欧盟的
GMP
检查依据的
IC
H
Q7A
的指导纲要,厂家要参照此指导进行自身检查;其次,所有的质量管理文件、操作
规范(
SOP
)和各种生产管理表格、标牌、标签和生产记录都应当具备中英文对照,能够
让国外的审查官员看懂;其三,要对员工进行
GMP
的全员培训,了解并适应国外检查的特
点。
COS
认证过程对企业是有积极意义的,会使企业的
GMP
管理达到国际水平,而且随
着美、欧、日三方协调的进一步发展,通过欧盟的
GMP
检查和
COS
认证最终有可能直接
进入美国和日本市场,
至少会使美国
FDA
的注册变得更为容易。
因此,
尽管目前
EDQM
还
没有对
COS
认证的申请人全部进行
GMP
检查,但中国的原料药生产厂家在提出
COS
认
证申请的同时为欧盟
GMP
检查做充分的准备是值得的。
出口到欧盟药品需要在国内批准了吗
第一部分 欧盟药品管理概述
第二部分 欧盟GMP基本要求
引言
基本要求Ⅰ:人用药品及兽药制剂生产质量管理规范
基本要求Ⅱ:原料药生产质量管理规范
第三部分 欧盟GMP附录
欧盟GMP附录1 无菌药品的生产
欧盟GMP附录2 人用生物制品的生产
欧盟GMP附录3 放射品生产’
欧盟GMP附录4 兽用非免疫药品的生产
欧盟GMP附录5 免疫类兽药制品的生产
欧盟GMP附录6 医用气体生产
欧盟GMP附录7 草药制剂的生产
欧盟GMP附录8 原辅包装材料的取样
欧盟GMP附录9 液剂、霜剂和油膏的生产
欧盟GMP附录10 定量吸人式气雾剂的生产
欧盟GMP附录11 计算机系统
欧盟GMP附录12 药品生产中电离辐射的应用
欧盟GMP附录13 临床试验用药的生产
欧盟GMP附录14 人血液或血浆制品的生产
欧盟GMP附录15 确认和验证
欧盟GMP附录16 药品放行责任人签发证书和放行批产品
欧盟GMP附录17 参数放行
欧盟GMP附录19 对照样品和留样
欧盟GMP附录20 质量风险管理
欧盟GMP术语
第四部分 欧盟GMP原文
Introduction
Part Ⅰ-Chapter I Quatity management
Part Ⅱ-Basci Requirements for Active Substances used as Starting Materials
Annex 1 Manufacture of Sterile Medicinal Products
Annex 2 Manufacture of Biological Medicinal Products for Human Use
Annex 3 Manufacture of Radio Phannaceuticals
Annex 4 Manufacture of Veterinary Medicinal Products other than Immunological Veterinary Medicinal Products
Annex 5 Manufacture of Immunological Veterinary Medicinal Products
Annex 6 Manufacture of Medicinal Gases
Annex 7 Manufacture of Herbal Medicinal Products
Annex 8 Sampling of Starting and Packaging Materials
Annex 9 Manufacture of Liquids, Creams and Ointments
Annex 10 Manufacture of Pressurised Metered Dose Aerosol Preparations for Inhalation
Annex 11 Computerised Systems
Annex 12 Use of Ionising Radiation in the Manufacture of Medicinal Products
Annex 13 Manufacture of Investigational Medicinal Products
Annex 14 Manufacture of Products derived from Human Blood or Human Plasma
Annex 15 Qualification and validation (July 2001)
Annex 16 Certification by a Qualified Person and Batch Release (July 2001)
Annex 17 Parametric Release (July 2001)
Annex 19 Reference and Retention Samples (December 2005)
Annex 20 Quality Risk Management
出口欧盟原料药证明文件办理大概要多久
出口到欧盟药品需要在国内批准。2011年6月欧盟发布了原料药新指令2011/62/EU,要求对进口到欧盟成员国的原料药,自2013年7月2日起,其生产企业必须取得出口国药品监管机构签发的证明文件。出口欧盟原料药证明文件由原料药生产企业所在地省级食品药品监督管理部门负责出具。
可在6个月之内办完。
CEP的有效期为5年,CEP持有者应在过期前6个月申请更新,如若企业能坚持不断更新档案资料,CEP将具有无限期的有效期。CEP持有者须及时通知客户有关变更内容,且将对应已修订的CEP提供给客户。CEP的修订类型主要包括通知、小变更、重大变更、更新(5年之后)、专论修订或条例变更后更新等。
目前,我国原料药出口至欧盟市场须通过欧洲药品质量管理局(简称EDQM)现场检查,获取CEP证书。据EDQM官方数据显示,截至2011年12月7日,我国原料药企业共获取367个CEP证书,其中,有效CEP证书335个,被取消、暂停及已过期的CEP证书32个,企业被暂停和吊销CEP证书的情况时有发生。2010年期间,EDQM在全球范围内对34家生产企业进行了现场核查,暂停了16个CEP证书,撤销了8个,检查中的不合格率非常高。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。