原料药出口情况-原料药出口流程
原料药出口不可以按照普通化工品出口,按照《药事管理与法规》来出口。
出口是指本国生产或加工的商品输往国外市场销售。出口贸易不仅要追求出口数量,金额的绝对量的增加,而且要努力提高经济效益,这样的出口发展才有积极意义。
出口欧盟原料药证明文件办理大概要多久
第一条 为规范蛋白同化制剂、肽类激素的进出口管理,根据《中华人民共和国药品管理法》、《中华人民共和国海关法》、《反兴奋剂条例》等法律、行政法规,制定本办法。第二条 国家对蛋白同化制剂、肽类激素实行进出口准许证管理。第三条 进口蛋白同化制剂、肽类激素,进口单位应当向所在地省、自治区、直辖市食品药品监督管理部门提出申请。第四条 进口供医疗使用的蛋白同化制剂、肽类激素,进口单位应当报送以下资料:
(一)药品进口申请表。
(二)购货合同或者订单复印件。
(三)《进口药品注册证》(或者《医药产品注册证》)(正本或者副本)复印件。
(四)进口单位的《药品经营许可证》、《企业法人营业执照》、《进出口企业资格证书》(或者《对外贸易经营者备案登记表》)、《组织代码证书》复印件;药品生产企业进口本企业所需原料药和制剂中间体(包括境内分包装用制剂),应当报送《药品生产许可证》、《企业法人营业执照》、《组织代码证书》复印件。
(五)《进口药品注册证》(或者《医药产品注册证》)持有者如委托其他公司代理出口其药品的,需提供委托出口函。
上述各类复印件应当加盖进口单位公章。第五条 因教学、科研需要而进口蛋白同化制剂、肽类激素的,进口单位应当报送以下资料:
(一)药品进口申请表;
(二)购货合同或者订单复印件;
(三)国内使用单位合法资质的证明文件、药品使用数量的测算依据以及使用单位出具的合法使用和管理该药品保证函;
(四)相应科研项目的批准文件或者相应主管部门的批准文件;
(五)接受使用单位委托代理进口的,还需提供委托代理协议复印件和进口单位的《企业法人营业执照》、《进出口企业资格证书》(或者《对外贸易经营者备案登记表》)、《组织代码证书》复印件。
上述各类复印件应当加盖进口单位公章。第六条 境内企业因接受境外企业委托生产而需要进口蛋白同化制剂、肽类激素的,报送本办法第五条第一款第(一)项、第(三)项、第(五)项规定的资料。
上述各类复印件应当加盖进口单位公章。第七条 省、自治区、直辖市食品药品监督管理部门收到进口申请及有关资料后,应当于15个工作日内作出是否同意进口的决定;对同意进口的,发给药品《进口准许证》;对不同意进口的,应当书面说明理由。第八条 进口蛋白同化制剂、肽类激素必须经由批准的允许药品进口的口岸进口。进口单位持省、自治区、直辖市食品药品监督管理部门核发的药品《进口准许证》向海关办理报关手续。进口蛋白同化制剂、肽类激素无需办理《进口药品通关单》。第九条 进口供医疗使用的蛋白同化制剂、肽类激素(包括首次在中国销售的),进口单位应当于进口手续完成后,及时填写《进口药品报验单》,持《进口药品注册证》(或者《医药产品注册证》)原件(正本或者副本)、药品《进口准许证》原件,向进口口岸食品药品监督管理部门报送下列资料一式两份,申请办理《进口药品口岸检验通知书》:
(一)《进口药品注册证》(或者《医药产品注册证》)(正本或者副本)和药品《进口准许证》复印件;
(二)进口单位的《药品生产许可证》或者《药品经营许可证》复印件,《企业法人营业执照》复印件;
(三)原产地证明复印件;
(四)购货合同复印件;
(五)装箱单、提运单和货运发票复印件;
(六)出厂检验报告书复印件;
(七)药品说明书及包装、标签的式样(原料药和制剂中间体除外)。
上述各类复印件应当加盖进口单位公章。第十条 口岸食品药品监督管理部门接到《进口药品报验单》及相关资料,审查无误后,将《进口药品注册证》(或者《医药产品注册证》)(正本或者副本)原件、药品《进口准许证》原件交还进口单位,并应当于当日向负责检验的口岸药品检验所发出《进口药品口岸检验通知书》,附本办法第九条规定的资料1份。
口岸药品检验所接到《进口药品口岸检验通知书》后,应当在2个工作日内与进口单位联系,到存货地点进行抽样,抽样完成后,应当在药品《进口准许证》原件第一联背面注明“已抽样”字样,并加盖抽样单位的公章。
国家对药品出口有什么管理制度和规定?
可在6个月之内办完。
CEP的有效期为5年,CEP持有者应在过期前6个月申请更新,如若企业能坚持不断更新档案资料,CEP将具有无限期的有效期。CEP持有者须及时通知客户有关变更内容,且将对应已修订的CEP提供给客户。CEP的修订类型主要包括通知、小变更、重大变更、更新(5年之后)、专论修订或条例变更后更新等。
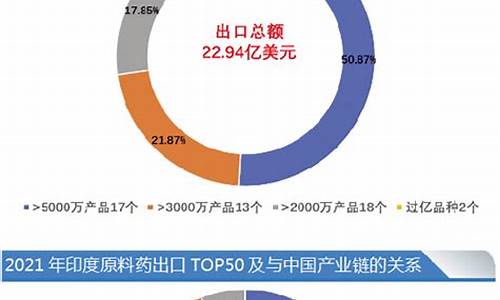
目前,我国原料药出口至欧盟市场须通过欧洲药品质量管理局(简称EDQM)现场检查,获取CEP证书。据EDQM官方数据显示,截至2011年12月7日,我国原料药企业共获取367个CEP证书,其中,有效CEP证书335个,被取消、暂停及已过期的CEP证书32个,企业被暂停和吊销CEP证书的情况时有发生。2010年期间,EDQM在全球范围内对34家生产企业进行了现场核查,暂停了16个CEP证书,撤销了8个,检查中的不合格率非常高。
一、2004年6月30日以前, 国所 药品制剂和原料药 生产必须符合GMP要求,并取得“药品GMP证书”。
二、生产血液制品、粉针剂和大容量注射剂、小容量注射剂企业,若 局分别规定 1998年12月31日(国药管安〔1998〕110号)、2000年12月31日(国药管安〔1999〕261号)、2002年12月31日(国药管安〔1999〕261号)后,仍未取得该剂型或类别“药品GMP 证书” ,一律不得生产该剂型或类别药品。生产其它剂型、类别药品 企业,自2004年7月1日起,凡未取得相应剂型或类别“药品GMP证书” ,一律停止其生产。 关《药品生产许可证》以及相应 药品生产批准文号,由药品监督管理部门依法处理。
三、凡申请药品GMP 认证 药品生产企业,应 2003年12月底前完成申报工作,并将相关资料报送所 地省、自治区、直辖市药品监督管理局。
四、自2003年1月1日起,药品生产企业若有未取得“药品GMP证书”的药品类别或剂型(包括生产车间、生产线),并准备申请药品GMP认证的,应一次性同时申报,我局将不再受理同一企业多次GMP认证申请。
五、体外诊断试剂、中药饮片、药用辅料、医用氧气、药用空心胶囊等生产企业应按GMP要求组织生产,其认证管理规定另行通知。
六、新开办药品生产企业(包括新增生产范围、新建生产车间)必须通过GMP认证,取得“药品GMP证书”后,方可生产。
七、申请仿制药品的生产企业,若未取得相应剂型或类别“药品GMP证书”,我局不受理其仿制药品生产申请。
八、申请新药生产的药品生产企业,若在我局规定的药品GMP认证期限后,仍未取得相应剂型或类别“药品GMP证书”,我局将不予核发其相应的药品生产批准文号。
九、凡未取得“药品GMP证书”的药品生产企业,一律不得接受相应剂型药品的委托生产。
十、药品经营企业和医疗机构在药品招标采购工作中,应优先选购取得“药品GMP证书”的药品生产企业生产的药品。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。