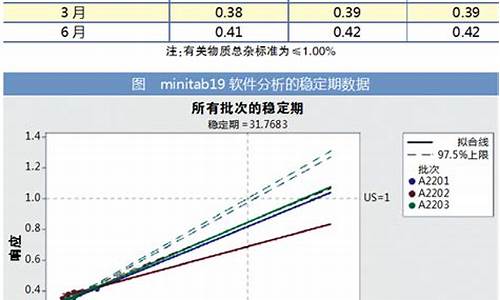
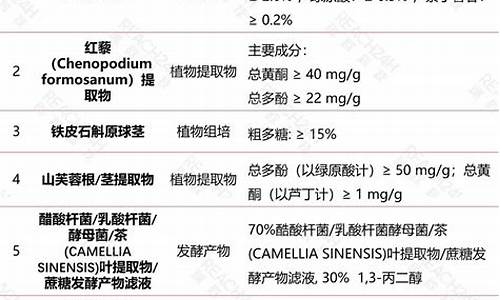
原料药含量限度以什么表示-原料药含量限度

原料药分析是指对于没有经过加工的药物原料的分析,工作主要是关于所用药物的分子式等有关的药物分子结构的分析,以及粒径大小等与其溶解度有关的性质分析,制剂分析中主要考虑的是制剂因素对于药物的溶解度以及溶出速度和生物利用度等有关的因素,不仅包括了药物本身的性质分析,也包括了剂型因素,处方设计,以及辅料应用和制备工艺的分析,不知道这么说楼主明白不
化妆品有毒吗?
没明白你的问题
你是想问:为什么做制剂的过程中,原料药中的杂质含量会增高?
还是为什么制剂的杂质含量限度比原料药高?
反正这么说吧,我不知道你是哪一种药物。但是一般的药物都会有一些不稳定的因素。比如光照、高温、高湿、酸、碱、氧化。在制剂过程中,有些是不能避免的。
比如你湿法制粒,那么肯定原料药会遇到水,然后烘干过程肯定会经过高温。诸如此类的步骤,导致了原料药中的杂质增高。所以一般来说,制剂的杂质比原料药的含量高一些属于正常现象。制剂的杂质限度也会比原料药的杂质限度宽松一些。
原料中控制的杂质,制剂中有必要或者必须继续控制吗?依据
化妆品一般都是乳剂或膏剂
制作过程中用到的原料药或一些辅料都是有国家标准的,前面的说含铅是不完全的,那要看化妆品的主要成分是什么和工艺中用了什么化学试剂。一般这些东西都有重金属和砷盐的含量限度,如<2ppm之类的,这些都是在安全范围之内的。有毒是不可能的,因为要是有毒的话这个产品是不会批生产的。
古代就有用含铅、水银、砒霜之类的东西美容,现在是不可能用的了。
新兽药及兽药新制剂管理办法
有,需要继续控制。
原料药是原料药,制剂是制剂。你知道这些杂质是原料药中带入的,只能证明了它的来源。现在对于杂质的要求非常严格,即便是原料药中的杂质也分为:起始物料、中间体或者降解产物等等。
看这个吧,超过了报告限度的杂质,是需要出报告的。超过鉴定限度的杂质是需要知道它具体的化学结构的。
尤其是你在制剂研究、生产过程中监控这些杂质的增长情况。有些杂质在原料药里增长不明显,做成了制剂就蹭蹭地长。到底是湿度影响,还是酸碱度影响,或者是温度影响。这关系到制剂工艺的问题,另外产品贮藏条件,包装条件都是跟主成分含量以及杂质有关系的。
药物分析笔记:药物分析的基础知识
第一章 总则第一条 根据《兽药管理条例》第二十二条、第二十三条及第二十四条的规定,制定本办法。第二条 新兽药系指我国新研制的兽药原料药品及其制剂。兽药新制剂系指用国家已批准的兽药原料药品新研制、加工出的兽药制剂。
已批准生产的兽药制剂,凡改变处方、剂型、给药途径和增加新的适应症的,亦属兽药新制剂。第三条 凡从事新兽药研究、生产、经营、检验、监督管理的单位和人员,都必须遵守本办法。第二章 新兽药及兽药新制剂的分类第四条 按管理要求,新兽药分以下五类:
第一类 我国创制的原料药品及其制剂(包括天然药物中提取的及合成的新发现的有效单体及其制剂);我国研制的国外未批准生产、仅有文献报道的原料药品及其制剂。
新发现的中药材;中药材新的药用部位。
第二类 我国研制的国外已批准生产,但未列入国家药典、兽药典或国家法定药品标准的原料药品及其制剂。
天然药物中提取的有效部分及其制剂。
第三类 我国研制的国外已批准生产,并已列入国家药典、兽药典或国家法定药品标准的原料药品及其制剂;天然药物中已知有效单体用合成或半合成方法制取的原料药品及其制剂。
西兽药复方制剂,中西兽药复方制剂。
第四类 改变剂型或改变给药途径的药品。
新的中药制剂(包括古方、秘方、验方、改变传统处方组成的);改变剂型但不改变给药途径的中成药。
第五类 增加适应症的西兽药制剂、中兽药制剂(中成药)。第五条 新兽药命名要明确、简短、科学,不准用代号及容易混同或夸大疗效的名称。第三章 新兽药及兽药新制剂的研制要求第六条 新兽药的研究内容应包括:理化性质、药理、毒理、临床、处方、剂量、剂型、稳定性、生产工艺等,并提出质量标准草案。第七条 新兽药临床药效试验,按照新兽药类别分为临床试验和临床验证。第一、二类新兽药必须进行临床试验;兽药新制剂必须进行临床验证;第三类新兽药做临床试验或临床验证,但必须经农业部认定。第八条 新兽药临床试验,根据研制的不同阶段,分为实验临床试验和扩大区域试验。
实验临床试验是用中间试制生产的3-5批产品,在小规模条件下研究新兽药对使用对象动物的药效和安全性做出试验结果和评价,必要时应进行人工感染模拟试验。
扩大区域试验是在自然生产条件下、较大范围内考察新兽药对使用对象动物的临床药效和安全性。第九条 实验临床试验的动物数目应不少于下列规定:
治疗药物 驱虫药物 饲料药物添加剂
大家畜 40头 60头 100头
中家畜 60头 100头 200头
小家畜及家禽 100只 300只 500只
鱼类 100尾 300尾 500尾
蜜蜂 10标准箱 20标准箱
蚕 10张 20张 40张
外用驱虫药物的试验动物数目应加倍第十条 实验临床试验应设对照组。对照组的动物应与试验动物条件一致。第十一条 第一、二、三类新兽药的实验临床试验应由农业部认可的省属或部属科研单位、高等院校、医疗单位承担;兽药新制剂应由省、自治区、直辖市畜牧(农牧)厅(局)认可的单位承担。第十二条 实验临床试验药品应由研制单位免费提供。在临床试验中因药品质量造成的不良后果,应由研制单位承担责任。第十三条 实验临床试验结束后,在经农业部批准试生产期内,须进行扩大区域试验。扩大区域试验的动物数目应不少于实验临床试验规定的动物数目的三倍至五倍。第十四条 临床验证主要考察新兽药或兽药新制剂的疗效和毒副反应,与原药品对照组进行对比验证。临床验证的试验动物数目,可以按第九条规定的数目减半。第四章 新兽药及兽药新制剂的审批第十五条 研制单位完成新兽药实验临床试验后,必须向所在省、自治区、直辖市畜牧(农牧)厅(局)提出新兽药试生产或生产申请,并按规定报送有关资料及样品。第一、二、三类新兽药由省、自治区、直辖市畜牧(农牧)厅(局)签署意见后报农业部审批;兽药新制剂由省、自治区、直辖市畜牧(农牧)厅(局)受理审批 。第十六条 申报新兽药,须提交下列内容资料:
(一)新兽药名称(包括正式品名、化学名、拉丁名、汉语拼音等,并说明命名依据)。
(二)选题的目的与依据,国内外有关该药研究现状或生产、使用情况的综述。
(三)新兽药的化学结构或组份的试验数据、理化常数、图谱及对图谱的解析。
(四)新兽药的合成路线、工艺条件、精制方法、原料和辅料的规格标准;动植物原料的来源、学名、药用或提取部位;抗生素的菌种来源、培养基的标准及配方;制剂的处方、处方依据和工艺。
(五)原料药及其制剂、复方制剂稳定性试验报告。
(六)药理学试验结果,包括作用机制、药代动力学试验及抑菌、消毒药的最小抑菌浓度试验等。
(七)毒理试验结果,包括实验动物和使用对象动物的急性、慢性毒性试验,局部用药的刺激性和吸收毒性试验等。
(八)特殊毒性试验,包括生殖毒性、致突变、致癌试验。
(九)机体残留试验及屠宰前停药期的研究报告。
(十)激素、饲料药物添加剂的动物传代繁育试验报告。
(十一)驱虫药、消毒药等外用药对环境毒性(植物毒性、水族毒性、昆虫毒性)研究及对土壤、水质污染的研究报告。
(十二)临床试验结果,包括实验临床试验、饲喂试验、药效学试验等。
(十三)中试生产的总结报告,中试生产的合成路线、工艺条件、精制方法、原料和辅料标准并与实验室制品的对比。
(十四)连续中试生产的样品3-5批及其检验报告书。送检样品量至少应为全检量的五倍。
(十五)三废处理试验报告。
(十六)质量标准草案及起草说明。主要内容包括:名称、结构式及分子式、含量限度、处方、理化性状、鉴别项目及方法和依据,含量(效价)测定的方法和依据、检查项目及方法和依据,标准品或化学对照品的来源及其制备方法、作用与用途、用法与用量、注意事项、制剂的规格、贮藏、有效期等。
(十七)新兽药及其制剂的包装、标签、使用说明书。
(十八)生产成本计算。
(十九)主要参考文献。试验结果与主要参考文献有不同的,应加以论证说明。
以上试验资料,按新兽药所属类别或用途不同而分别提供(详见附件)。
原料和制剂的细菌内毒素限度需要一致吗
药品检验工作的基本程序:
一般为取样、鉴别、检查、含量测定、写出报告。
取样:
鉴别:判断真伪。 检查:称纯度检查,判定药物优劣。 含量测定:测定药物中有效成分的含量。检验报告必须明确、肯定、有依据。计量仪器认证要求:县级以上人民政府计量行政部门负责进行监督检查。符合经济合理、就地就近。
药品质量标准分析方法验证:
目的是证明采用的方法适合于相应的检测要求。 验证内容:准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐用性。
一、准确度: 是指用该方法测定的结果与真实值或参考值接近的程度,一般以百分回收率表示。至少用9次测定结果进行评价。[医学教育 网 搜集整理]
二、精密度: 是指在规定的条件下,同一个均匀样品,经过多次取样测定所得结果之间的接近程度。用偏差、标准偏差或相对标准偏差表示。
1、 重复
2、 性:相同
3、 条件下,
4、 一个分析人员测定所得结果的精密度称为重复
5、 性。至少9次。
6、 中间精密度:
7、 一个实验室,
8、 不同
9、 时间不同
10、 分析人员用不同
11、 设备
12、 测定结果的精密度。
3、重现性:不同实验室,不同分析人员测定结果的精密度。分析方法被法定标准采用应进行重现性试验。
三、专属性:指在其他成分可能存在的情况下,采用的方法能准确测定出被测物的特性,用于复杂样品分析时相互干扰的程度。鉴别反应、杂质检查、含量测定方法,圴应考察专属性。
四、检测限:指试样中被测物能被检测出的最低量,无须定量。用百分数、ppm或ppb表示。
五、定量限:指样品中被测物能被定量测定的最低量,测定结果应具一定的精密度和准确度。
六、线性:系指在设计的范围内,测试结果与试样中被测物浓度直接呈正比关系的程度。
七、范围:能达到一定的精密度、准确度和线性的条件下,测试方法适用的高低限浓度或量的区间。 八、耐用性:指在一定的测定条件稍有变动时,测定结果不受影响的承受程度。
药物分析的统计学知识
测量误差:测量值和真实值之差。 绝对误差和相对误差。真实值:是有经验的人用最可靠的方法对试样进行多次测定所得的平均值。
系统误差:(1)方法误差 (2)试剂误差 (3)仪器误差 (4)操作误差偶然误差:不可定误差或随机误差,由偶然原因引起。可增加平得测定次数。 测量值的准确度表示测量的正确性,测量值的精密度表示测量的重现性。精密度是表示准确度的先决条件,只有在消除了系统误差后,才可用精密度同时表达准确 度。
提高分析准确度方法:
1、选择合适的分析方法
2、减少测量误差
3、增加平行测定次数
4、消除测量过程中的系统误差(校准仪器、做对照试验、做回收试验、做空白试验)有效数字的处理:0.05060g是四位有效数字。首位是8或9,有效 数字可多记一位。ph=8.02是两位有效数字。四舍六入五成双原则。修约标准偏差或其他表示不确定度时,修约结果可使准确度估计值变得差一点。s= 2.13——2.2 g检验法、4d法,>舍去。
药品质量标准制定的原则和基本内容
原则:安全有效,技术先进,经济合理。 检验方法:准确、灵敏、简便、快速。
(一)、名称:
(二)、性状:
1、外观、臭、味和稳定性
2、溶解度:一定程度上反映药品的纯度。
3、物理常数
(1)馏程:2000规定:在标准压力(101.3kpa)下,按药典装置,自开始馏出的第五滴算起,至供试品仅剩3-4ml或一定比例的容积馏出时的温度范围。
(2)熔点:系指一种物质固体熔化成液体的温度,熔融同时分解的温度,或在熔化时自初熔至全熔的一段温度。
(3)凝点:系指一种物质由液体凝结为固体时,在短时间内停留不变的温度。
(4)比旋度:具光学异构体分子的药物,旋光性能不同。按干燥品或无水物计算。准确至0.01 .
(5)折光率:光线自一种透明介质进入另一种透明介质时,两种介质密度不同,光的进行速度发生变化,即发生折射现象,遵从折射定律。对于液体药品,尤其是植物油,检查药品的纯杂程度,测定溶液的浓度。
(6)粘度:流体对流动的阻抗能力。共三法,毛细管内径。
(7)吸收系数:物质对光的选择性吸收波长。[医学教育网 搜集整 理]
(三) 鉴别:用理化方法或生物学方法来证明药品真实性的方法。对已知物。
(四) 杂质检查:有效性,纯度要求和安全性。
1、有效性试验
2、酸碱度
3、溶液的澄清度与颜色
4、无机阴离子:氯化物和硫酸盐。
5、有机杂质
6、干燥失重和水分
7、炽灼残渣:指硫酸化灰分,用于考察有机药物中混入的无机杂质。一般限度为0.1% .
8、金属离子和重金属检查 每日剂量0.5g以上且长期服用的品种。
9、硒和砷:硒检查有:醋酸地塞米松、醋酸曲安奈德及醋酸氟轻松。第一法:古蔡氏法 10、安全性检查
(五)含量测定或效价测定: 理化方法称含量测定 生物学方法或生化方法测定称效价测定。
1、 容量分析法:化学原料药含量测定的首选法。中和法、非水滴定法、银量法、络合法、碘量法、重氮化法。
2、 重量法:精密度好准确度高,
3、 繁琐,
4、 不
5、 能应用容量法时用。挥发法、萃取法、沉淀法
6、 紫外分光光度法:简便、快速。原料药避免。
7、 气相色谱法:分离效果优越,
8、 对含杂质和挥发性的原料药效好。维生素e
9、 高效液相色谱法:用于多组分抗生素,
10、 生化药品或因杂质干扰测定。常规方法又难分离药品。
原料药和新制剂中的杂质,哪些需要对其定性或结构确证
有,需要继续控制。原料药是原料药,制剂是制剂。你知道这些杂质是原料药中带入的,只能证明了它的来源。现在对于杂质的要求非常严格,即便是原料药中的杂质也分为:起始物料、中间体或者降解产物等等。看这个吧,超过了报告限度的杂质,是需要出报告的。超过鉴定限度的杂质是需要知道它具体的化学结构的。尤其是你在制剂研究、生产过程中监控这些杂质的增长情况。有些杂质在原料药里增长不明显,做成了制剂就蹭蹭地长。到底是湿度影响,还是酸碱度影响,或者是温度影响。这关系到制剂工艺的问题,另外产品贮藏条件,包装条件都是跟主成分含量以及杂质有关系的。
原料药是原料药,制剂是制剂。你知道这些杂质是原料药中带入的,只能证明了它的来源。现在对于杂质的要求非常严格,即便是原料药中的杂质也分为:起始物料、中间体或者降解产物等等。
看这个吧,超过了报告限度的杂质,是需要出报告的。超过鉴定限度的杂质是需要知道它具体的化学结构的。
尤其是你在制剂研究、生产过程中监控这些杂质的增长情况。有些杂质在原料药里增长不明显,做成了制剂就蹭蹭地长。到底是湿度影响,还是酸碱度影响,或者是温度影响。这关系到制剂工艺的问题,另外产品贮藏条件,包装条件都是跟主成分含量以及杂质有关系的。,具体的可以查询药智网,回答满意请您采纳,谢谢。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。