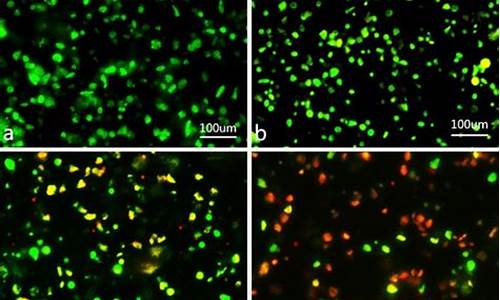
简述原料药或制剂生产过程的有害因素-原料药和制剂的连续制造方法
第一章 总则第一条 根据《兽药管理条例》第二十二条、第二十三条及第二十四条的规定,制定本办法。第二条 新兽药系指我国新研制的兽药原料药品及其制剂。兽药新制剂系指用国家已批准的兽药原料药品新研制、加工出的兽药制剂。
已批准生产的兽药制剂,凡改变处方、剂型、给药途径和增加新的适应症的,亦属兽药新制剂。第三条 凡从事新兽药研究、生产、经营、检验、监督管理的单位和人员,都必须遵守本办法。第二章 新兽药及兽药新制剂的分类第四条 按管理要求,新兽药分以下五类:
第一类 我国创制的原料药品及其制剂(包括天然药物中提取的及合成的新发现的有效单体及其制剂);我国研制的国外未批准生产、仅有文献报道的原料药品及其制剂。
新发现的中药材;中药材新的药用部位。
第二类 我国研制的国外已批准生产,但未列入国家药典、兽药典或国家法定药品标准的原料药品及其制剂。
天然药物中提取的有效部分及其制剂。
第三类 我国研制的国外已批准生产,并已列入国家药典、兽药典或国家法定药品标准的原料药品及其制剂;天然药物中已知有效单体用合成或半合成方法制取的原料药品及其制剂。
西兽药复方制剂,中西兽药复方制剂。
第四类 改变剂型或改变给药途径的药品。
新的中药制剂(包括古方、秘方、验方、改变传统处方组成的);改变剂型但不改变给药途径的中成药。
第五类 增加适应症的西兽药制剂、中兽药制剂(中成药)。第五条 新兽药命名要明确、简短、科学,不准用代号及容易混同或夸大疗效的名称。第三章 新兽药及兽药新制剂的研制要求第六条 新兽药的研究内容应包括:理化性质、药理、毒理、临床、处方、剂量、剂型、稳定性、生产工艺等,并提出质量标准草案。第七条 新兽药临床药效试验,按照新兽药类别分为临床试验和临床验证。第一、二类新兽药必须进行临床试验;兽药新制剂必须进行临床验证;第三类新兽药做临床试验或临床验证,但必须经农业部认定。第八条 新兽药临床试验,根据研制的不同阶段,分为实验临床试验和扩大区域试验。
实验临床试验是用中间试制生产的3-5批产品,在小规模条件下研究新兽药对使用对象动物的药效和安全性做出试验结果和评价,必要时应进行人工感染模拟试验。
扩大区域试验是在自然生产条件下、较大范围内考察新兽药对使用对象动物的临床药效和安全性。第九条 实验临床试验的动物数目应不少于下列规定:
治疗药物 驱虫药物 饲料药物添加剂
大家畜 40头 60头 100头
中家畜 60头 100头 200头
小家畜及家禽 100只 300只 500只
鱼类 100尾 300尾 500尾
蜜蜂 10标准箱 20标准箱
蚕 10张 20张 40张
外用驱虫药物的试验动物数目应加倍第十条 实验临床试验应设对照组。对照组的动物应与试验动物条件一致。第十一条 第一、二、三类新兽药的实验临床试验应由农业部认可的省属或部属科研单位、高等院校、医疗单位承担;兽药新制剂应由省、自治区、直辖市畜牧(农牧)厅(局)认可的单位承担。第十二条 实验临床试验药品应由研制单位免费提供。在临床试验中因药品质量造成的不良后果,应由研制单位承担责任。第十三条 实验临床试验结束后,在经农业部批准试生产期内,须进行扩大区域试验。扩大区域试验的动物数目应不少于实验临床试验规定的动物数目的三倍至五倍。第十四条 临床验证主要考察新兽药或兽药新制剂的疗效和毒副反应,与原药品对照组进行对比验证。临床验证的试验动物数目,可以按第九条规定的数目减半。第四章 新兽药及兽药新制剂的审批第十五条 研制单位完成新兽药实验临床试验后,必须向所在省、自治区、直辖市畜牧(农牧)厅(局)提出新兽药试生产或生产申请,并按规定报送有关资料及样品。第一、二、三类新兽药由省、自治区、直辖市畜牧(农牧)厅(局)签署意见后报农业部审批;兽药新制剂由省、自治区、直辖市畜牧(农牧)厅(局)受理审批 。第十六条 申报新兽药,须提交下列内容资料:
(一)新兽药名称(包括正式品名、化学名、拉丁名、汉语拼音等,并说明命名依据)。
(二)选题的目的与依据,国内外有关该药研究现状或生产、使用情况的综述。
(三)新兽药的化学结构或组份的试验数据、理化常数、图谱及对图谱的解析。
(四)新兽药的合成路线、工艺条件、精制方法、原料和辅料的规格标准;动植物原料的来源、学名、药用或提取部位;抗生素的菌种来源、培养基的标准及配方;制剂的处方、处方依据和工艺。
(五)原料药及其制剂、复方制剂稳定性试验报告。
(六)药理学试验结果,包括作用机制、药代动力学试验及抑菌、消毒药的最小抑菌浓度试验等。
(七)毒理试验结果,包括实验动物和使用对象动物的急性、慢性毒性试验,局部用药的刺激性和吸收毒性试验等。
(八)特殊毒性试验,包括生殖毒性、致突变、致癌试验。
(九)机体残留试验及屠宰前停药期的研究报告。
(十)激素、饲料药物添加剂的动物传代繁育试验报告。
(十一)驱虫药、消毒药等外用药对环境毒性(植物毒性、水族毒性、昆虫毒性)研究及对土壤、水质污染的研究报告。
(十二)临床试验结果,包括实验临床试验、饲喂试验、药效学试验等。
(十三)中试生产的总结报告,中试生产的合成路线、工艺条件、精制方法、原料和辅料标准并与实验室制品的对比。
(十四)连续中试生产的样品3-5批及其检验报告书。送检样品量至少应为全检量的五倍。
(十五)三废处理试验报告。
(十六)质量标准草案及起草说明。主要内容包括:名称、结构式及分子式、含量限度、处方、理化性状、鉴别项目及方法和依据,含量(效价)测定的方法和依据、检查项目及方法和依据,标准品或化学对照品的来源及其制备方法、作用与用途、用法与用量、注意事项、制剂的规格、贮藏、有效期等。
(十七)新兽药及其制剂的包装、标签、使用说明书。
(十八)生产成本计算。
(十九)主要参考文献。试验结果与主要参考文献有不同的,应加以论证说明。
以上试验资料,按新兽药所属类别或用途不同而分别提供(详见附件)。
原料药的药物制剂
原料药是我国发明的词汇,在国外其不能称之为“正式的药品”,而是药品活性成分!
原料药药厂主要应用化工合成或者应用生物技术来合成这些原料药(还有一部分天然药物提取),然后交付给制剂药厂,经过添加辅料制成普通制剂(注射剂、片剂、胶囊、口服液、外用制剂。。。)以及缓控释制剂、靶向制剂等。只有制作成制剂药品,才能真正应用于广大人民群众。
原料药与药物制剂的质量控制在方法的选择上有什么异同?
具体的原料药加工后——药物制剂
原料药的称呼主要相对于制剂来说的。
以化学加工手段获得的原料为主,供应生产成品药的原料
比如注射用硫酸头孢匹罗是药,那么硫酸头孢匹罗就是原料药
原料和制剂的细菌内毒素限度需要一致吗
在药典上可以清晰的看出,由于制剂可能有辅料的干扰,所以一般尽量用高效液相来进行含量测定,而原料则可以用滴定法或紫外、荧光等来检测含量。但对于那些杂质紫外吸收和药品紫外吸收波长相同的原料也要用高效液相色谱法。
有,需要继续控制。原料药是原料药,制剂是制剂。你知道这些杂质是原料药中带入的,只能证明了它的来源。现在对于杂质的要求非常严格,即便是原料药中的杂质也分为:起始物料、中间体或者降解产物等等。看这个吧,超过了报告限度的杂质,是需要出报告的。超过鉴定限度的杂质是需要知道它具体的化学结构的。尤其是你在制剂研究、生产过程中监控这些杂质的增长情况。有些杂质在原料药里增长不明显,做成了制剂就蹭蹭地长。到底是湿度影响,还是酸碱度影响,或者是温度影响。这关系到制剂工艺的问题,另外产品贮藏条件,包装条件都是跟主成分含量以及杂质有关系的。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。