吖啶橙荧光染料进行细胞染色-吖啶橙染色液
RNA与DNA最重要的区别一是RNA只有一条链,二是它的碱基组成与DNA的不同,RNA没有碱基T(胸腺嘧啶),而有碱基U(尿嘧啶)。所以导致他们有以下性质上的不同。
1.两性解离:DNA无,只有酸解离,碱基被屏蔽(在分子内部形成了H键)。RNA有,有PI。
2.粘度大:DNA;RNA,粘度由分子长度/直径决定,DNA为线状分子,RNA为线团。
3.碱的作用:DNA耐碱RNA易被碱水解。
4.显色反应:
鉴别DNA和RNA+浓HCl RNA ------→ 绿色化合物
DNA ------→ 蓝紫色化合物苔黑酚
二苯胺啡啶溴红(荧光染料)和溴嘧啶都可对DNA染色,原理是卡在分子中,DNA的离心和电泳显色可用它们。
DNA和RNA的鉴别染色
利用吖啶橙的变色特性可鉴别DNA和RNA。吖啶橙作为一种荧光染料已被用于染色固定,非固定细胞核酸,或作溶酶体的一种标记。观察亡细胞荧光变色性变化以及区别分裂细胞和静止细胞群体。虽然测定DNA和RNA含量时较难获得好的重复性结果,但该方法已被许多实验室广泛采用。
5.溶解性:都溶于水而不溶于乙醇,因此,常用乙醇来沉淀溶液中的DNA和RNA。DNA溶于苯酚而RNA不溶,故可用苯酚来沉淀RNA。
6.紫外吸收:核酸的λm=260nm,碱基展开程度越大,紫外吸收就越厉害。当A=1时,DNA:50ug/ml,RNA和单链DNA:40ug/ml,寡核苷酸:20ug/ml。用A260/A280还可来表示核酸的纯度。
7.沉降速度:对于拓扑异构体(核苷酸数目相同的核酸),其沉降速度从达到小依次为:RNA ; 超螺旋DNA > 解链环状DNA ; 松弛环状DNA ; 线形DNA也就是在离心管中最上层是线形DNA,最下面是RNA。
8.电泳:核苷酸、核酸均可以进行电泳,泳动速度主要由分子大小来决定,因此,电泳是测定核酸分子量的好方法。
9.DNA分子量测定最直接的方法:用适当浓度的EB(溴嘧啶)染色DNA,可以将其他形式的DNA变成线形DNA,用电镜测出其长度,按B-DNA模型算出bp数,根据核苷酸的平均分子量就可计算出DNA的分子量。
关于细胞生物学实验的问题!!!!
碘化丙啶(propiolium iodide,PI)能嵌入DNA双螺旋中,可使荧光强度增加约20倍,以488nm波长激发,DNA/PI复合物最大的发射波长约为615nm。
小鼠Lewis肺癌细胞DNA含量测定方法
(1).从C57BL/6小鼠上切除肿块,在培养皿内用PBS冲洗。
(2).去除结缔组织及,剪碎肿块。
(3).小碎片移入1.20×38mm针,加压使其通过,于4℃条件下重悬细胞于HBSS中。
(4).将200~300μL细胞悬液(5×105细胞/mL)中加入3mL PI(50μg/mL),染色3LL细胞,于4℃存放20~30分钟。
(5).测定580~750nm之间的发射荧光,以去除末结合PI产生的激发光与发射光谱线之间的重叠部分。
注:PI染色液:0.1%柠檬酸钠1000mL+PI 5mg + 1% Nonide P40水。
2. 培养细胞DNA的流式细胞仪分析
(1).从培养皿中吸去培养基,以HBSS冲洗二次。
(2).加入PI5mL于培养皿中,在4℃放10分钟。
(3).用吸管反复次打细胞,使细胞破坏,胞核释放出来,再行流式细胞仪分析。
3. 完整细胞DNA的PI染色
(1).70%乙醇固定的细胞悬液,离心,去固定液。
(2).室温条件下加入PI染色一批细胞(105~106细胞/mL),时间为30分钟,然后行流式细胞仪分析。
(二)吖啶橙染色
1. DNA和RNA的鉴别染色
利用吖啶橙的变色特性可鉴别DNA和RNA。吖啶橙作为一种荧光染料已被用于染色固定,非固定细胞核酸,或作溶酶体的一种标记。观察亡细胞荧光变色性变化以及区别分裂细胞和静止细胞群体。虽然测定DNA和RNA含量时较难获得好的重复性结果,但该方法已被许多实验室广泛采用。方法如下:
1)试剂:
(1)溶液A:低温保存,稳定期约2周。
Triton X-100(0.1%) 0.1mL,1mol/L HCL 8mL?
1mol/L NaCL 15ml蒸馏水76mL,P
荧光显微镜的使用方法
实验、叶绿体的分离与荧光观察
实 验 目 的
1. 通过植物细胞叶绿体的分离, 了解细胞器分离的一般原理和方法。
2. 观察叶绿体的自发荧光和次生荧光, 并熟悉荧光显微镜的使用方法。
实 验 原 理
将组织匀浆后悬浮在等渗介质中进行差速离心,是分离细胞器的常用方法。叶绿体的分离应在等渗溶液(0.35mol/L氯化钠或0.4mol/L蔗糖溶液)中进行, 以免渗透压的改变使叶绿体到损伤。将匀浆液1000r/min的条件下离心2min, 以去除其中的组织残渣和未被破碎的完整细胞。然后,在3000r/min的条件下离心5min,即可获得沉淀的叶绿体(混有部分细胞核)。分离过程最好在0~5℃的条件下进行;如果在室温下,要迅速分离和观察。
利用荧光显微镜对可发荧光的物质进行检测时,将受到许多因素的影响,如温度、光、淬灭剂等。因此在荧光观察时应抓紧时间, 有必要时立即拍照。另外,在制作荧光显微标本时最好使用无荧光载片、盖片和无荧光油。
实 验 用 品
一、器材
1.主要设备: 普通离心机、组织捣碎机、粗天平、荧光显微镜。
2.小型器材: 500ml烧杯2个, 250ml量筒1个, 滴管20支, 10ml刻度离心管20支, 试管架5个,纱布若干,无荧光载片和盖片各4片。
二、材料 新鲜菠菜。
三、试剂 0.35mol/L氯化钠溶液,0.01%吖啶橙(acridine orange)。
实 验 方 法
一、叶绿体的分离与观察
1. 选取新鲜的嫩菠菜叶,洗净擦干后去除叶梗脉,称30g于150ml 0.35mol/L NaCl溶液中,装入组织捣碎机。
2. 利用组织捣碎机低速(5000r/min)匀桨3~5min。
3. 将匀浆用6层纱布过滤于500ml烧杯中。
4. 取滤液4ml在1000r/min下离心2min。弃去沉淀。
5. 将上清液在3000r/min下离心5min。弃去上清液,沉淀即不叶绿体(混有部分细胞核)。
6. 将沉淀用0.35mol/LNaCl溶液悬浮、
7. 取叶绿体悬液一滴滴于载玻片上,加盖玻片后即可在普通光镜和荧光显微镜下观察。
(1)在普通光镜下观察。
(2)在荧光显微镜下观察叶绿体的直接荧光。
(3)在荧光显微镜下观察叶绿体的间接荧光:取叶绿体悬液一滴滴在无荧光载片上,再滴加一滴0.01%吖啶橙荧光染料, 加盖片后即可在荧光显微镜下观察。
二、菠菜叶手切片观察
用剔须刀片将新鲜的嫩菠菜叶切削出一斜面置于载玻片上,滴加1~2滴0.35mol/L NaCl溶液,加盖片后轻压,置显微镜下观察。
(1)在普通光镜下观察。
(2)在荧光显微镜下观察其直接荧光。
(3) 观察间接荧光:向手切片上滴加1~2滴0.01%吖啶橙染液,染色1min,洗去余液, 加盖片后可在荧光显微镜下观察间接荧光。
实 验 结 果
一、叶绿体的分离和观察
1. 普通光镜下,可看到叶绿体为绿色橄榄形,在高倍镜下可看到叶绿体内部含有较深的绿色小颗粒,即基粒。
2. 以Olympus荧光显微镜为例,在先用B(bule)激发滤片、B双色镜和O530(orange)阴断滤片的条件下,叶绿体发出火红色荧光。
3. 加入吖啶橙染色后,叶绿体可发出桔红色荧光,而其中混有的细胞核则发绿色荧光。
二、菠菜叶手切片观察
1. 在普通光镜下可以看到三种细胞 (1)表皮细胞: 为边缘呈锯齿形的鳞片状细胞; (2)保卫细胞: 为构成气孔的成对存在的肾形细胞;(3)叶肉细胞: 为排列成栅状的长形和椭圆形细胞。叶绿体呈绿色橄榄形,在高倍镜下还可以看到绿色的基粒。
2. 在荧光显微镜下,叶绿体发出火红色荧光,但其荧光强度要比游离叶绿体弱, 气孔发绿色荧光,两保卫细胞内的火红色叶绿体则环绕气孔排列成一圈。表皮细胞内的叶绿体数量要比叶肉细胞少。
3. 用吖啶橙染色后,叶绿体则发出桔红色荧光,细胞核可发出绿色荧光, 气孔仍为绿色。
作 业
1. 在普通光学显微镜下,用目微尺和台微尺测量一下叶绿体的长轴和短轴,分别测量5~10个叶绿体,求其平均值。
2. 在荧光显微镜下,观察叶绿体的自发荧光时,更换滤镜系统,叶绿体的颜色是否有变化?
实验五、 植物染色体标本制备与观察
实验目的
学习植物染色体标本的制备技术,了解Feulgen反应的基本原理,学习其操作方法和压片法,初步掌握细胞分裂各期的主要特点.
核酸是生物最重要的组成成分,核酸分为两大类,即脱氧核糖核酸(DNA)和核糖核酸(RNA).它们在细胞内的分布及化学性质均有所不同.DNA主要分布在核的染色质或有丝分裂过程中出现的染色体内.RNA分布在细胞质和核仁内.但在染色质及染色体中也含有少量的RNA,核仁中也有少量的DNA.在线粒体,叶绿体等细胞器内除含有RNA外,也含有少量的DNA.
实验原理
Feulgen反应的原理是根据DNA经弱酸(1mol/L)水解后,打开料嘌呤和脱氧核糖连接的键,在脱氧核糖的一端形成有离的醛基.这些醛基就在原位与Schiff试剂结合,形成含醌基( )的化合物,醛基是一个发色团所以具有颜色,因此凡有DNA的部位,就呈现紫红色.
实验材料
大麦、黑麦或小麦种子
实验内容
植物染色体标本的制备及DNA的显示法-Feulgen反应
压片法
细胞有丝分裂相的观察
实验步骤
(一)植物染色体标本的制备
1、将植物种子放在潮湿的滤纸上,20°C发芽,待胚根长至1~2cm时,切取0.5cm长的根尖部分。
2、预处理:将切下的根尖浸入0.1%秋水酰素液中,室温下处理3~4小时。
3、固定、水解及染色:
Camoy固定剂
固定10-30分钟 ? 95%酒精10分钟 ? 70%酒精10分钟 ? 蒸馏水 →(对照) 5%三氯醋酸 ? 90oC 15分钟
1mol/L HCl ← 蒸馏水 ? 1mol/L HCl ? 60 oC 8分钟 ? Schiff试剂40 -60分钟 ? 亚硫酸水(1,2,3) ? 换3次,每次5分钟自来水 ? 将染色后的根尖漂洗干净,悬着染色效果好的材料 ? 蒸馏水 ? 压片 ? 镜检(二) 压片法
压片的操作步骤如下,把根尖放在载片上,用刀片切取根尖(0.3cm),纵切根尖,加一滴水,用解剖针将根尖纵向分成若干小条,保留1-2小条,加盖玻片,用铅笔端轻轻敲击盖玻片,使细胞分离,压平呈云雾状.
(三)蚕豆(或洋葱)根尖压片的观察
首先在低倍镜下,区分根尖的分生组织区及延长组织区的细胞,然后,在高倍镜下,观察细胞核的形态,注意在你的片子上,是否有有丝分裂细胞,如有,你能找到细胞分裂期的几个阶段,其特征如何 (可参照附录)
做Feulgen反应时,应注意的几个问题
固定剂的选择:一般常选用Carnoy固定液.其他的固定剂如Flemming固定剂及Champy固定剂等均可用,但不能使用Bouin固定剂.
水解时间:水解时间一定要合适,不宜过长或不足,否则会影响实验结果.如用Carnoy固定液固定的材料,水解时间一般在8-15分钟之间.水解时间的长短要随不同的材料及不同的固定剂而定.
Schiff试剂的质量:在做Feulgen反应时,重要的因素就是Schiff试剂的质量问题.实验时,要注意试剂颜色是否正常,有无SO2的气味.
洗涤剂的重要性:漂洗时,所用的亚硫酸水,最好在每次实验前临时配制,以便保持较浓的SO2.
实验对照组:一定要做对照片,以便说明实验结果的真实性.
操作过程中,用镊子镊取根尖的生长区部位,切勿夹取根冠部位.
鸦片过程中尽量使根尖分生组织细胞保持原来的分布状态.
习题
简述Feulgen反应的原理
欲得到一张好的Feulgen反应制片,制片过程中应注意些什么
绘制细胞分裂图。
实验六 植物原生质体的制备
实验目的
1.学习植物原生质体的分离制备技术,观察原生质体的形态。
2.学习植物原生质体的融合技术,观察不同方法融合细胞的形态及变化。
实验用品
显微镜,擦镜纸,剪子,镊子,小平皿,吸管四支,直式漏斗,300目尼龙网,10ml离心管,载片,盖片。
实验原理
原生质体(protoplast)这个术语最早是由Hanstein在1880年提出来的。确切地说,食用菌原生质体是指细胞壁完全消除后余下的那部分包裹的裸露的细胞结构。
植物的花瓣细胞在经过纤维素酶,离折酶处理后,纤维素和果胶成分遭到破坏,造成原生质体脱离细胞壁进入培养液中。
植物花瓣细胞中的大液泡中存在有大量色素,且不同的细胞色素不同,因此可以在不对细胞染色的情况下,通过颜色范围辨认不同细胞的原生质体,以及同种间细胞融合和异种间细胞融合情况。
PEG是一种高分子化合物,由于含有醚键而具有负极性,与水,蛋白质和碳水化合物等一些正极化基团形成氢键。当PEG分子足够长时,可作为邻近原生质体表面之间的分子桥而使之粘连。PEG也能连接Ca2+等阳离子。Ca2+可在一些负极化基团和PEG之间形成桥,因而促进粘连,在洗涤过程中,连接在原生质体膜上的PEG分子可被洗脱,这将引起电荷的紊乱和再分布,从而引起原生质体融合,高Ca2+高pH清洗则增加了质膜的流动性,因而大大提高了融合频率,洗涤时的渗透冲击对融合也可能起作用。
实验试剂
1.洗涤液:
甘露醇 0.6 M
CaCl2?2H2O 8 mM
NaH2PO4?H2O 2 Mm
Ph 5.6
实验步骤
1.酶液的制备 把纤维素酶(EAS-867)溶于0.5~0.7摩尔/升甘露醇内,加10毫摩/升氯化钙溶液作稳定剂。酶溶解后,经4000转/分离心机沉降原生质体,20分钟后,取上清液。经针筒型过滤器,再经0.45微米微孔滤膜过滤后,在无菌箱内把酶液分装在三角烧瓶内,贮存在冰箱里备用。
2.材料准备 把生长在温室,苗龄60天以上的烟草植株,取上中部全展叶,在日光下照射2小时,使它萎蔫。也可以用温室生长的蚕豆叶作同样处理,或用大田收获的胡萝卜,放在0℃以上低温备用。萎蔫后的叶片浸在3%的漂白精片溶液中15~20分钟,再用无菌水冲洗干净,用无菌吸水纸吸干表面水分。
3.酶解 用尖头镊子撕去叶片下表皮,剪成小块。胡萝卜则用刀片削去表皮和中柱,取用皮层部,切成小块。以上材料1克,放入盛有10毫升酶溶液的培养皿里,加盖。在28~30℃下保温1.5~3小时。
4.洗涤 叶片或其他组织经酶解后会出现圆形的游离原生质体,原生质体悬浮液内有未消化的组织、碎片、细胞和破裂的原生质体。用滤纸或尼龙布滤去粗杂质,再把原生质体悬浮液移到离心管,用手摇离心机转动2分钟,吸去上清液(见图),再加入0.5毫升甘露醇洗涤2~3次,最后就得到较为纯净的原生质体,可以供进一步培养或作其他实验用。
实验七 细胞融合方法
实验目的
1、初步掌握动物细胞PEG融合的方法。
2、学习细胞融合及其应用的有关知识。
实验原理
2个或2个以上的细胞合并为1个细胞的现象,称为细胞融合。细胞融合的主要方法有病毒法、聚乙二醇(PEG)法和电融合方法。
用PEG处理细胞,能使质膜性质发生改变,导致细胞膜融汇,胞质流通,最后导致细胞融合。
实验器材、材料与试剂
1、仪器:
光学显微镜,离心机,恒温水浴锅,C02培养箱,超净台,倒置显微镜等。
2、材料
新鲜鸡红细胞,无菌注射器,6号针头,刻度离心管,试管,载玻片,盖玻片。
3、试剂
50%PEG,Hanks液,甲醇,Giemsa染液,碘酒,75%乙醇
实验方法
(1)取新鲜鸡血以0.85%生理盐水制成10%的悬液。
(2)称取0.5克PEG(MW=4,000)放入试管内,在酒精灯上融化之,迅速加入0.5ml预热的Hanks液混匀制成50%的PEG溶液。放入37℃水浴中待用。
(3)取上述10%的鸡红细胞悬液1ml放入离心管中,再加入5ml Hanks液混匀,然后以1,000rpm离心5分钟,小心弃去上清,用指弹法将细胞团块弹散。
(4)取上述50%PEG溶液0.5ml,在1分钟内滴加到鸡红细胞悬液,边加边轻轻摇动混匀。待PEG全部加入后静置1分钟左右。此全部过程都要求在37℃水浴内进行。
(5)缓慢滴加9ml Hanks液以终止PEG的作用,在37℃水浴内静置5分钟。
(6)离心弃上清后,取一滴融合后的细胞悬液滴片,加盖片镜检。
实验室中常用的DNA分子量的测定方法有哪些
(1)打开灯源,超高压汞灯要预热15min才能达到最亮点。
(2)透射式荧光显微镜需在光源与暗视野聚光器之间装上所要求的激发滤片,在物镜的后面装上相应的压制滤片。落射式荧光显微镜需在光路的插槽中插入所要求的激发滤片、双色束分离器、压制滤片的插块。
(3)用低倍镜观察,根据不同型号荧光显微镜的调节装置,调整光源中心,使其位于整个照明光斑的中央。
(4)放置标本片,调焦后即可观察。使用中应注意:未装滤光片不要用眼直接观察,以免引起眼的损伤;用油镜观察标本时,必须用无荧光的特殊镜油;高压汞灯关闭后不能立即重新打开,需待汞灯完全冷却后才能再启动,否则会不稳定,影响汞灯寿命。
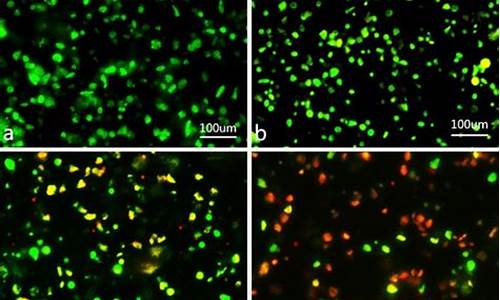
(5)观察。例如:在荧光显微镜下用蓝紫光滤光片,观察到经0.01%吖啶橙荧光染料染色的细胞,细胞核和细胞质被激发产生两种不同颜色的荧光(暗绿色和橙红色)。
请详细解释一下DNA与RNA
第一种方法最常用
1)紫外吸收法也就是测量OD(260)和OD(280)的吸收值,这样的方法其它的杂质对测量结果影响大一点,因为其它杂质在这两个吸收波长也有吸收.
(2)荧光法,用PicoGreen荧光染料,测定DNA,RNA浓度比较灵敏,并且适合测量低浓度和微量DNA和RNA,并且受其它杂质的影响不大,缺点要有专门的荧光检测仪器,试剂比较昂贵.
纯化DNA可以买试剂盒,主要有膜吸附法,磁珠分离法,都很方便.
RNA与DNA最重要的区别一是RNA只有一条链,二是它的碱基组成与DNA的不同,RNA没有碱基T(胸腺嘧啶),而有碱基U(尿嘧啶).所以导致他们有以下性质上的不同.
1.两性解离:DNA无,只有酸解离,碱基被屏蔽(在分子内部形成了H键).RNA有,有PI.
2.粘度大:DNA;RNA,粘度由分子长度/直径决定,DNA为线状分子,RNA为线团.
3.碱的作用:DNA耐碱RNA易被碱水解.
4.显色反应:
鉴别DNA和RNA+浓HCl RNA ------→ 绿色化合物
DNA ------→ 蓝紫色化合物苔黑酚
二苯胺啡啶溴红(荧光染料)和溴嘧啶都可对DNA染色,原理是卡在分子中,DNA的离心和电泳显色可用它们.
DNA和RNA的鉴别染色
利用吖啶橙的变色特性可鉴别DNA和RNA.吖啶橙作为一种荧光染料已被用于染色固定,非固定细胞核酸,或作溶酶体的一种标记.观察亡细胞荧光变色性变化以及区别分裂细胞和静止细胞群体.虽然测定DNA和RNA含量时较难获得好的重复性结果,但该方法已被许多实验室广泛采用.
5.溶解性:都溶于水而不溶于乙醇,因此,常用乙醇来沉淀溶液中的DNA和RNA.DNA溶于苯酚而RNA不溶,故可用苯酚来沉淀RNA.
6.紫外吸收:核酸的λm=260nm,碱基展开程度越大,紫外吸收就越厉害.当A=1时,DNA:50ug/ml,RNA和单链DNA:40ug/ml,寡核苷酸:20ug/ml.用A260/A280还可来表示核酸的纯度.
7.沉降速度:对于拓扑异构体(核苷酸数目相同的核酸),其沉降速度从达到小依次为:RNA ; 超螺旋DNA > 解链环状DNA ; 松弛环状DNA ; 线形DNA也就是在离心管中最上层是线形DNA,最下面是RNA.
8.电泳:核苷酸、核酸均可以进行电泳,泳动速度主要由分子大小来决定,因此,电泳是测定核酸分子量的好方法.
9.DNA分子量测定最直接的方法:用适当浓度的EB(溴嘧啶)染色DNA,可以将其他形式的DNA变成线形DNA,用电镜测出其长度,按B-DNA模型算出bp数,根据核苷酸的平均分子量就可计算出DNA的分子量.
荧光染料的科研应用
RNA与DNA最重要的区别一是RNA只有一条链,二是它的碱基组成与DNA的不同,RNA没有碱基T(胸腺嘧啶),而有碱基U(尿嘧啶)。所以导致他们有以下性质上的不同。
1.两性解离:DNA无,只有酸解离,碱基被屏蔽(在分子内部形成了H键)。RNA有,有PI。
2.粘度大:DNA;RNA,粘度由分子长度/直径决定,DNA为线状分子,RNA为线团。
3.碱的作用:DNA耐碱RNA易被碱水解。
4.显色反应:
鉴别DNA和RNA+浓HCl RNA ------→ 绿色化合物
DNA ------→ 蓝紫色化合物苔黑酚
二苯胺啡啶溴红(荧光染料)和溴嘧啶都可对DNA染色,原理是卡在分子中,DNA的离心和电泳显色可用它们。
DNA和RNA的鉴别染色
利用吖啶橙的变色特性可鉴别DNA和RNA。吖啶橙作为一种荧光染料已被用于染色固定,非固定细胞核酸,或作溶酶体的一种标记。观察亡细胞荧光变色性变化以及区别分裂细胞和静止细胞群体。虽然测定DNA和RNA含量时较难获得好的重复性结果,但该方法已被许多实验室广泛采用。
5.溶解性:都溶于水而不溶于乙醇,因此,常用乙醇来沉淀溶液中的DNA和RNA。DNA溶于苯酚而RNA不溶,故可用苯酚来沉淀RNA。
6.紫外吸收:核酸的λm=260nm,碱基展开程度越大,紫外吸收就越厉害。当A=1时,DNA:50ug/ml,RNA和单链DNA:40ug/ml,寡核苷酸:20ug/ml。用A260/A280还可来表示核酸的纯度。
7.沉降速度:对于拓扑异构体(核苷酸数目相同的核酸),其沉降速度从达到小依次为:RNA ; 超螺旋DNA > 解链环状DNA ; 松弛环状DNA ; 线形DNA也就是在离心管中最上层是线形DNA,最下面是RNA。
8.电泳:核苷酸、核酸均可以进行电泳,泳动速度主要由分子大小来决定,因此,电泳是测定核酸分子量的好方法。
9.DNA分子量测定最直接的方法:用适当浓度的EB(溴嘧啶)染色DNA,可以将其他形式的DNA变成线形DNA,用电镜测出其长度,按B-DNA模型算出bp数,根据核苷酸的平均分子量就可计算出DNA的分子量。
聚合酶链反应(Polymerase Chain Reaction ,PCR)是80年代中期发展起来的体外核酸扩增技术。它具有特异、敏感、产率高、快速、简便、重复性好、易自动化等突出优点;能在一个试管内将所要研究 的目的基因或某一DNA片段于数小时内扩增至十万乃至百万倍,使肉眼能直接观察和判断;可从一根毛发、一滴血、甚至一个细胞中扩增出足量的DNA供分析研 究和检测鉴定。过去几天几星期才能做到的事情,用PCR几小时便可完成。PCR技术是生物医学领域中的一项革命性创举和里程碑。
PCR技术简史
PCR的最早设想 核酸研究已有100多年的历史,本世纪60年代末、70年代初人们致力于研究基因的体外分离技术,Korana于1971年最早提出核酸体外扩增的设想:“经过DNA变性,与合适的引物杂交,用DNA聚合酶延伸引物,并不断重复该过程便可克隆tRNA基因”。
PCR的实现 1985年美国PE-Cetus公司人类遗传研究室的Mullis等发明了具有划时代意义的聚合酶链反应。其原理类似于DNA的体内复制,只是在试管中给 DNA的体外合成提供以致一种合适的条件---摸板DNA,寡核苷酸引物,DNA聚合酶,合适的缓冲体系,DNA变性、复性及延伸的温度与时间。
PCR的改进与完善 Mullis最初使用的DNA聚合酶是大肠杆菌DNA聚合酶I的 Klenow片段,其缺点是:①Klenow酶不耐高温,90℃会变性失活,每次循环都要重新加。②引物链延伸反应在37℃下进行,容易发生模板和引物之 间的碱基错配,其PCR产物特异性较差,合成的DNA片段不均一。此种以Klenow酶催化的PCR技术虽较传统的基因扩增具备许多突出的优点,但由于 Klenow酶不耐热,在DNA模板进行热变性时,会导致此酶钝化,每加入一次酶只能完成一个扩增反应周期,给PCR技术操作程序添了不少困难。这使得 PCR技术在一段时间内没能引起生物医学界的足够重视。1988年初,Keohanog改用T4 DNA聚合酶进行PCR,其扩增的DNA片段很均一,真实性也较高,只有所期望的一种DNA片段。但每循环一次,仍需加入新酶。1988年Saiki 等从温泉中分离的一株水生嗜热杆菌(thermus aquaticus) 中提取到一种耐热DNA聚合酶。此酶具有以下特点:①耐高温,在70℃下反应2h后其残留活性大于原来的90%,在93℃下反应2h后其残留活性是原来的 60%,在95℃下反应2h后其残留活性是原来的40%。②在热变性时不会被钝化,不必在每次扩增反应后再加新酶。③大大提高了扩增片段特异性和扩增效 率,增加了扩增长度(2.0Kb)。由于提高了扩增的特异性和效率,因而其灵敏性也大大提高。为与大肠杆菌多聚酶I Klenow片段区别,将此酶命名为Taq DNA多聚酶(Taq DNA Polymerase)。此酶的发现使PCR广泛的被应用。
PCR技术基本原理
PCR技术的基本原理 类似于DNA的 天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性--退火--延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加 热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板DNA与引 物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板--引物结合 物在TaqDNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA 链互补的半保留复制链重复循环变性--退火--延伸三过程,就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需 2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。到达平台期(Plateau)所需循环次数取决于样品中模板的拷贝。
PCR的反应动力学 PCR的三个反应步骤反复进行,使DNA扩增量呈指数上升。反应最终的DNA 扩增量可用Y=(1+X)n计算。Y代表DNA片段扩增后的拷贝数,X表示平(Y)均每次的扩增效率,n代表循环次数。平均扩增效率的理论值为100%, 但在实际反应中平均效率达不到理论值。反应初期,靶序列DNA片段的增加呈指数形式,随着PCR产物的逐渐积累,被扩增的DNA片段不再呈指数增加,而进 入线性增长期或静止期,即出现“停滞效应”,这种效应称平台期数、PCR扩增效率及DNA聚合酶PCR的种类和活性及非特异性产物的竟争等因素。大多数情 况下,平台期的到来是不可避免的。
PCR扩增产物 可分为长产物片段和短产物片段两部分。短产物片段的长度严格地限定在两个引物链5’端之间,是需要扩增的特定片段。短产物片段和长产物片段是由于引物所 结合的模板不一样而形成的,以一个原始模板为例,在第一个反应周期中,以两条互补的DNA为模板,引物是从3’端开始延伸,其5’端是固定的,3’端则没 有固定的止点,长短不一,这就是“长产物片段”。进入第二周期后,引物除与原始模板结合外,还要同新合成的链(即“长产物片段”)结合。引物在与新链结合 时,由于新链模板的5’端序列是固定的,这就等于这次延伸的片段3’端被固定了止点,保证了新片段的起点和止点都限定于引物扩增序列以内、形成长短一致的 “短产物片段”。不难看出“短产物片段”是按指数倍数增加,而“长产物片段”则以算术倍数增加,几乎可以忽略不计, 这使得PCR的反应产物不需要再纯化,就能保证足够纯DNA片段供分析与检测用。
PCR反应体系与反应条件
标准的PCR反应体系:
10×扩增缓冲液 10ul
4种dNTP混合物 各200umol/L
引物 各10~100pmol
模板DNA 0.1~2ug
Taq DNA聚合酶 2.5u
Mg2+ 1.5mmol/L
加双或三蒸水至 100ul
PCR反应五要素: 参加PCR反应的物质主要有五种即引物、酶、dNTP、模板和Mg2+
引物: 引物是PCR特异性反应的关键,PCR 产物的特异性取决于引物与模板DNA互补的程度。理论上,只要知道任何一段模板DNA序列,就能按其设计互补的寡核苷酸链做引物,利用PCR就可将模板DNA在体外大量扩增。
设计引物应遵循以下原则:
①引物长度: 15-30bp,常用为20bp左右。
②引物扩增跨度: 以200-500bp为宜,特定条件下可扩增长至10kb的片段。
③引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C过多易出现非特异条带。ATGC最好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列。
④避免引物内部出现二级结构,避免两条引物间互补,特别是3’端的互补,否则会形成引物二聚体,产生非特异的扩增条带。
⑤引物3’端的碱基,特别是最末及倒数第二个碱基,应严格要求配对,以避免因末端碱基不配对而导致PCR失败。
⑥引物中有或能加上合适的酶切位点,被扩增的靶序列最好有适宜的酶切位点,这对酶切分析或分子克隆很有好处。
⑦引物的特异性:引物应与核酸序列数据库的其它序列无明显同源性。
引物量: 每条引物的浓度0.1~1umol或10~100pmol,以最低引物量产生所需要的结果为好,引物浓度偏高会引起错配和非特异性扩增,且可增加引物之间形成二聚体的机会。
酶及其浓度 目前有两种Taq DNA聚合酶供应, 一种是从栖热水生杆菌中提纯的天然酶,另一种为大肠菌合成的基因工程酶。催化一典型的PCR反应约需酶量2.5U(指总反应体积为100ul时),浓度过高可引起非特异性扩增,浓度过低则合成产物量减少。
dNTP的质量与浓度 dNTP的质量与浓度和PCR扩增效率有密切关系,dNTP粉呈颗粒状,如保存不当易变性失去生物学活性。dNTP溶液呈酸性,使用时应配成高浓度后,以1M NaOH或1M Tris。HCL的缓冲液将其PH调节到7.0~7.5,小量分装, -20℃冰冻保存。多次冻融会使dNTP降解。在PCR反应中,dNTP应为50~200umol/L,尤其是注意4种dNTP的浓度要相等( 等摩尔配制),如其中任何一种浓度不同于其它几种时(偏高或偏低),就会引起错配。浓度过低又会降低PCR产物的产量。dNTP能与Mg2+结合,使游离的Mg2+浓度降低。
模板(靶基因)核酸 模板核酸的量与纯化程度,是PCR成败与否的关键环节之一,传统的DNA纯化方法通常采用SDS和蛋白酶K来消化处理标本。 SDS的主要功能是: 溶解细胞膜上的脂类与蛋白质,因而溶解膜蛋白而破坏细胞膜,并解离细胞中的核蛋白,SDS 还能与蛋白质结合而沉淀;蛋白酶K能水解消化蛋白质,特别是与DNA结合的组蛋白,再用有机溶剂酚与氯仿抽提掉蛋白质和其它细胞组份,用乙醇或异丙醇沉淀 核酸。提取的核酸即可作为模板用于PCR反应。一般临床检测标本,可采用快速简便的方法溶解细胞,裂解病原体,消化除去染色体的蛋白质使靶基因游离,直接 用于PCR扩增。RNA模板提取一般采用异硫氰酸胍或蛋白酶K法,要防止RNase降解RNA。
Mg2+浓度 Mg2+对PCR扩增的特异性和产量有显著的影响,在一般的PCR反应中,各种dNTP浓度为200umol/L时,Mg2+浓度为1.5~2.0mmol/L为宜。Mg2+浓度过高,反应特异性降低,出现非特异扩增,浓度过低会降低Taq DNA聚合酶的活性,使反应产物减少。
PCR反应条件的选择
PCR反应条件为温度、时间和循环次数。
温度与时间的设置: 基于PCR原理三步骤而设置变性-退火-延伸三个温度点。在标准反应中采用三温度点法,双链DNA在90~95℃变性,再迅速冷却至40 ~60℃,引物退火并结合到靶序列上,然后快速升温至70~75℃,在Taq DNA 聚合酶的作用下,使引物链沿模板延伸。对于较短靶基因(长度为100~300bp时)可采用二温度点法, 除变性温度外、退火与延伸温度可合二为一,一般采用94℃变性,65℃左右退火与延伸(此温度Taq DNA酶仍有较高的催化活性)。
①变性温度与时间:变性温度低,解链不完全是导致PCR失败的最主要原因。一般情况下,93℃~94℃lmin足以使模板DNA变性,若低于93℃则 需延长时间,但温度不能过高,因为高温环境对酶的活性有影响。此步若不能使靶基因模板或PCR产物完全变性,就会导致PCR失败。
②退火(复性)温度与时间:退火温度是影响PCR特异性的较重要因素。变性后温度快速冷却至40℃~60℃,可使引物和模板发生结合。由于模板DNA 比引物复杂得多,引物和模板之间的碰撞结合机会远远高于模板互补链之间的碰撞。退火温度与时间,取决于引物的长度、碱基组成及其浓度,还有靶基序列的长 度。对于20个核苷酸,G+C含量约50%的引物,55℃为选择最适退火温度的起点较为理想。引物的复性温度可通过以下公式帮助选择合适的温度:
Tm值(解链温度)=4(G+C)+2(A+T)
复性温度=Tm值-(5~10℃)
在Tm值允许范围内, 选择较高的复性温度可大大减少引物和模板间的非特异性结合,提高PCR反应的特异性。复性时间一般为30~60sec,足以使引物与模板之间完全结合。
③延伸温度与时间:Taq DNA聚合酶的生物学活性:
70~80℃ 150核苷酸/S/酶分子
70℃ 60核苷酸/S/酶分子
55℃ 24核苷酸/S/酶分子
高于90℃时, DNA合成几乎不能进行。
PCR反应的延伸温度一般选择在70~75℃之间,常用温度为72℃,过高的延伸温度不利于引物和模板的结合。PCR延伸反应的时间,可根据待扩增片段的长度而定,一般1Kb以内的DNA片段,延伸时间1min是足够 的。3~4kb的靶序列需3~4min;扩增10Kb需延伸至15min。延伸进间过长会导致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。
循环次数 循环次数决定PCR扩增程度。PCR循环次数主要取决于模板DNA的浓度。一般的循环次数选在30~40次之间,循环次数越多,非特异性产物的量亦随之增多。
PCR反应特点
特异性强 PCR反应的特异性决定因素为:
①引物与模板DNA特异正确的结合;
②碱基配对原则;
③Taq DNA聚合酶合成反应的忠实性;
④靶基因的特异性与保守性。
其中引物与模板的正确结合是关键。引物与模板的结合及引物链的延伸是遵循碱基配对原则的。聚合酶合成反应的忠实性及Taq DNA聚合酶耐高温性,使反应中模板与引物的结合(复性)可以在较高的温度下进行,结合的特异性大大增加,被扩增的靶基因片段也就能保持很高的正确度。再通过选择特异性和保守性高的靶基因区,其特异性程度就更高。
灵敏度高 PCR产物的生成量是以指数方式增加的,能将皮克(pg=10-12g)量级的起始待测模板扩增到微克(ug=10-6g)水平。能从100万个细胞中检出一个靶细胞;在病毒的检测中,PCR的灵敏度可达3个RFU(空斑形成单位);在细菌学中最小检出率为3个细菌。
简便、快速 PCR反应用耐高温的Taq DNA聚合酶,一次性地将反应液加好后,即在DNA扩增液和水浴锅上进行变性-退火-延伸反应,一般在2~4 小时完成扩增反应。扩增产物一般用电泳分析,不一定要用同位素,无放射性污染、易推广。
对标本的纯度要求低 不需要分离病毒或细菌及培养细胞,DNA 粗制品及总RNA均可作为扩增模板。可直接用临床标本如血液、体腔液、洗嗽液、毛发、细胞、活组织等粗制的DNA扩增检测。 PCR扩增产物分析
PCR产物是否为特异性扩增 ,其结果是否准确可靠,必须对其进行严格的分析与鉴定,才能得出正确的结论。PCR产物的分析,可依据研究对象和目的不同而采用不同的分析方法。
凝胶电泳分析:PCR产物电泳,EB溴乙锭染色紫外仪下观察,初步判断产物的特异性。PCR产物片段的大小应与预计的一致,特别是多重PCR,应用多对引物,其产物片断都应符合预讦的大小,这是起码条件。
琼脂糖凝胶电泳: 通常应用1~2%的琼脂糖凝胶,供检测用。
聚丙烯酰胺凝胶电泳:6~10%聚丙烯酰胺凝胶电泳分离效果比琼脂糖好,条带比较集中,可用于科研及检测分析。
酶切分析:根据PCR产物中限制性内切酶的位点,用相应的酶切、电泳分离后,获得符合理论的片段,此法既能进行产物的鉴定,又能对靶基因分型,还能进行变异性研究。
分子杂交:分子杂交是检测PCR产物特异性的有力证据,也是检测PCR 产物碱基突变的有效方法。
Southern印迹杂交: 在两引物之间另合成一条寡核苷酸链(内部寡核苷酸)标记后做探针,与PCR产物杂交。此法既可作特异性鉴定,又可以提高检测PCR产物的灵敏度,还可知其分子量及条带形状,主要用于科研。
斑点杂交: 将PCR产物点在硝酸纤维素膜或尼膜薄膜上,再用内部寡核苷酸探针杂交,观察有无着色斑点,主要用于PCR产物特异性鉴定及变异分析。
现在常用的核酸染料有哪几种
荧光染料,由于灵敏度高,操作方便,逐渐取代了放射性同位素作为检测标记,其广泛应用于荧光免疫,荧光探针,细胞染色等。包括特异性的DNA染色,用于染色体分析、细胞周期、细胞凋亡等相关研究。另有很多核酸染料在多色染色系统中是非常有用的复染剂,可作为背景对照,标记细胞核使细胞内结构的空间关系一目了然。 核酸荧光染料对细胞核染色后定量测量细胞所发出的荧光强度,就可以确定细胞核中DNA、RNA的含量,并可以对细胞周期和细胞的增殖状况进行分析。有多种荧光染料可以对细胞中的DNA或RNA染色,常用的DNA染料包括碘化丙啶(PI)、DAPI、Hoechst 33342等,RNA染料有噻唑橙、吖啶橙等。
常用的核酸染料有:
1、EB染料
EB属于核酸分子嵌入剂,通常用于分子遗传学、DNA和染色质结构分析等研究中,特别是国内很多实验室仍在使用EB进行凝胶核酸电泳实验的染色。
EB是极易渗透细胞膜与胞内DNA嵌合的小分子,它具有平面共轭大环结构,是典型的DNA分子插入试剂,菲啶环插入到DNA分子的碱基对之间,与DNA嵌合形成稳定的复合物,并影响DNA
的复制,破坏正常的遗传生理现象。EB作为诱变性的化合物,它在人体中诱导突变的机制是不可逆转的。
2、GoldView染料
GoldView(GV)是一种可代替溴化乙锭(EB)的新型核酸染料,其灵敏度与EB相当,使用方法与之完全相同。在紫外透射光下双链DNA呈现绿色荧光,也可用于RNA染色。
实质上,所谓的Goldview就是吖啶橙,也就是传说中的AO,有一种传统的细胞凋亡试验,采用的染料正是吖啶橙,也就是AO/EB染色试验,EB无法穿透完整的细胞膜,而AO可以穿过细胞膜染上细胞核,以此来区分凋亡和未凋亡的细胞。
3、GelRed 和 GelGreen染料
GelRed
和GelGreen
是两种集高灵敏、低毒性和超稳定于一身的极佳的荧光核酸凝胶染色试剂。其水溶染色剂通过美国环保局安全认定,废弃物可直接倒入下水道,而不会造成任何环境污染。
GelRed
和 GelGreen
的特殊化学结构使其难以穿透细胞膜进入细胞,正是这一特性降低了染料的细胞毒性。不过也正是因为如此,其价格大约是GoldView的十倍左右。
扩展资料
核酸染料的科研应用:
1、免疫分析
荧光标记的单克隆抗体技术为流式细胞仪在研究细胞膜和细胞内各种功能性抗原、肿瘤基因蛋白等领域扩展了无限的应用空间。
荧光探针可以通过蛋白质交联剂共价结合在单克隆抗体上。免疫荧光标记最常用的染料有异硫氰酸荧光素(fluorescein isothiocyanate, FITC)、藻红蛋白(PE)以及AlexaFluor系列染料等。
2、核酸检测
核酸荧光染料对细胞核染色后定量测量细胞所发出的荧光强度,就可以确定细胞核中DNA、RNA的含量,并可以对细胞周期和细胞的增殖状况进行分析。
有多种荧光染料可以对细胞中的DNA或RNA染色,常用的DNA染料包括碘化丙啶(PI)、DAPI、Hoechst 33342等,RNA染料有噻唑橙、吖啶橙等。
百度百科-荧光染料
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。