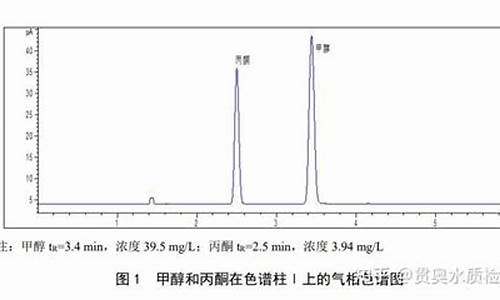
三溴丙酮气相检测方法有哪些要求-三溴丙酮气相检测方法有哪些要求和标准
有机物是有机化合物的简称,所有的有机物都含有碳元素。但是并非所有含碳的化合物都是有机化合物,比如CO,CO2。除了碳元素外有机物还可能含有其他几种元素。如H、N、S等。虽然组成有机物的元素就那么几种(碳最重要),但到现在人类却已经发现了超过1000万中有机物。而它们的特性更是千变万化。因此,有机化学是化学中一个相当重要的研究范畴。
有机物即碳氢化合物(烃)及其衍生物,简称有机物。除水和一些无机盐外,生物体的组成成分几乎全是有机物,如淀粉、蔗糖、油脂、蛋白质、核酸以及各种色素。过去误以为只有动植物(有机体)能产生有机物,故取名“有机”。现在不仅许多天然产物可以用人工方法合成,而且可以从动植物、煤、石油、天然气等分离或改造加工制成多种工农业生产和人民生活的必需品,象塑料、合成纤维、农药、人造橡胶等。与无机物相比,有机物的种类众多,一般挥发性较大、熔点和沸点较低,反应较慢(较复杂)。溶于有机溶剂,且能燃烧。碳原子可用共价键彼此连接生成多种结构,组成数量巨大的不同种类的有机分子骨架。按照基本结构,有机物可分成3类:(1)开链化合物,又称脂肪族化合物,因为它最初是在油脂中发现的。其结构特点是碳与碳间连接成不闭口的链。(2)碳环化合物(含有完全由碳原子组成的环),又可分成脂环族化合物(在结构上可看成是开链化合物关环而成的)和芳香族化合物(含有苯环)两个亚类。(3)杂环化合物(含有由碳原子和其他元素组成的环)。在烃分子中,共价连接的碳原子是骨架,碳的其他键则与氢结合。烃骨架非常稳定,因为形成碳-碳单键和双键的碳原子同等享用它们之间的电子对。烃的氢原子可以被不同的功能团(官能团)取代产生不同类的有机物。功能团决定分子的主要性质,所以有机物也常根据其功能团分类。有机生物分子的功能团比其烃骨架在化学上活泼得多,它们能改变邻近原子的几何形状及其上的电子分布,从而改变整个有机分子的化学反应性。从有机分子中的功能团可以分析和推测其化学行为和反应。如酶(细胞的催化剂)可识别生物分子中的特殊功能团并催化其结构发生特征性变化,大多数生物分子是多功能的,含有两种或多种功能团。在这些分子中,每种类型的功能团有其自己的化学特征和反应。如氨基酸具有至少两种功能团——氨基和羧基。丙氨酸的化学性质就基本决定于其氨基和羧基。又如葡萄糖也是多功能的生物分子,其化学性质基本决定于羟基和醛基两种功能团。生物分子的功能团在其生物活性中起着重要的作用。生物分子中某些其他的功能团列于下表中。
例如甲烷
甲烷分子式CH4。最简单的有机化合物。甲烷是没有颜色、没有气味的气体,沸点-161.4℃,比空气轻,它是极难溶于水的可燃性气体。甲烷和空气成适当比例的混合物,遇火花会发生爆炸。化学性质相当稳定,跟强酸、强碱或强氧化剂(如KMnO4)等一般不起反应。在适当条件下会发生氧化、热解及卤代等反应。
甲烷在自然界分布很广,是天然气、沼气、坑气及煤气的主要成分之一。它可用作燃料及制造氢、一氧化碳、炭黑、乙炔、氢氰酸及甲醛等物质的原料。
413kJ/mol、109°28′,甲烷分子是正四面体空间构型,上面的结构式只是表示分子里各原子的连接情况,并不能真实表示各原子的空间相对位置。
1.物质的理化常数:
国标编号 21007
CAS号 74-82-8
中文名称 甲烷
英文名称 methane;Marsh gas
别 名 沼气
分子式 CH4 外观与性状 无色无臭气体
分子量 16.04 蒸汽压 53.32kPa/-168.8℃ 闪点:-188℃
熔 点 -182.5℃ 沸点:-161.5℃ 溶解性 微溶于水,溶于醇、
密 度 相对密度(水=1)0.42(-164℃);相对密度(空气=1)0.55 稳定性 稳定
危险标记 4(易燃液体) 主要用途 用作燃料和用于炭黑、氢、乙炔、甲醛等的制造
2.对环境的影响:
一、健康危害
侵入途径:吸入。
健康危害:甲烷对人基本无毒,但浓度过高时,使空气中氧含量明显降低,使人窒息。当空气中甲烷达25%-30%时,可引起头痛、头晕、乏力、注意力不集中、呼吸和心跳加速、共济失调。若不及时脱离,可致窒息亡。皮肤接触液化本品,可致冻伤。
二、毒理学资料及环境行为
毒性:属微毒类。允许气体安全地扩散到大气中或当作燃料使用。有单纯性窒息作用,在高浓度时因缺氧窒息而引起中毒。空气中达到25~30%出现头昏、呼吸加速、运动失调。
急性毒性:小鼠吸入42%浓度×60分钟,麻醉作用;兔吸入42%浓度×60分钟,麻醉作用。
危险特性:易燃,与空气混合能形成爆炸性混合物,遇热源和明火有燃烧爆炸的危险。与五氧化溴、氯气、次氯酸、三氟化氮、液氧、二氟化氧及其它强氧化剂接触剧烈反应。
燃烧(分解)产物:一氧化碳、二氧化碳。
3.现场应急监测方法:
4.实验室监测方法:
气相色谱法《空气中有害物质的测定方法》(第二版),杭士平编
可燃溶剂所显色法;容量分析法《水和废水标准检验法》第20版(美)
5.环境标准:
前苏联 车间空气中有害物质的最高容许浓度 300mg/m3
美国 车间卫生标准 窒息性气体
6.应急处理处置方法:
一、泄漏应急处理
迅速撤离泄漏污染区人员至上风处,并进行隔离,严格限制出入。切断火源。建议应急处理人员戴自给正压式呼吸器,穿消防防护服。尽可能切断泄漏源。合理通风,加速扩散。喷雾状水稀释、溶解。构筑围堤或挖坑收容产生的大量废水。如有可能,将漏出气用排风机送至空旷地方或装设适当喷头烧掉。也可以将漏气的容器移至空旷处,注意通风。漏气容器要妥善处理,修复、检验后再用。
二、防护措施
呼吸系统防护:一般不需要特殊防护,但建议特殊情况下,佩带自吸过滤式防毒面具(半面罩)。
眼睛防护:一般不需要特别防护,高浓度接触时可戴安全防护眼镜。
身体防护:穿防静电工作服。
手防护:戴一般作业防护手套。
其它:工作现场严禁吸烟。避免长期反复接触。进入罐、限制性空间或其它高浓度区作业,须有人监护。
三、急救措施
皮肤接触:若有冻伤,就医治疗。
吸入:迅速脱离现场至空气新鲜处。保持呼吸道通畅。如呼吸困难,给输氧。如呼吸停止,立即进行人工呼吸。就医。
灭火方法:切断气源。若不能立即切断气源,则不允许熄灭正在燃烧的气体。喷水冷却容器,可能的话将容器从火场移至空旷处。灭火剂:雾状水、泡沫、二氧化碳、干粉。
第一部分:化学品名称
化学品中文名称:甲烷
化学品英文名称:methane
中文名称2:沼气
英文名称2:Marshgas
技术说明书编码:51
CASNo.:74-82-8
分子式:CH4
分子量:16.04
第二部分:成分/组成信息
有害物成分含量CASNo.
甲烷74-82-8
第三部分:危险性概述
危险性类别:
侵入途径:
健康危害:甲烷对人基本无毒,但浓度过高时,使空气中氧含量明显降低,使人窒息。当空气中甲烷达25%~30%时,可引起头痛、头晕、乏力、注意力不集中、呼吸和心跳加速、共济失调。若不及时脱离,可致窒息亡。皮肤接触液化本品,可致冻伤。
环境危害:
燃爆危险:本品易燃,具窒息性。
第四部分:急救措施
皮肤接触:若有冻伤,就医治疗。
眼睛接触:
吸入:迅速脱离现场至空气新鲜处。保持呼吸道通畅。如呼吸困难,给输氧。如呼吸停止,立即进行人工呼吸。就医。
食入:
第五部分:消防措施
危险特性:易燃,与空气混合能形成爆炸性混合物,遇热源和明火有燃烧爆炸的危险。与五氧化溴、氯气、次氯酸、三氟化氮、液氧、二氟化氧及其它强氧化剂接触剧烈反应。
有害燃烧产物:一氧化碳、二氧化碳。
灭火方法:切断气源。若不能切断气源,则不允许熄灭泄漏处的火焰。喷水冷却容器,可能的话将容器从火场移至空旷处。灭火剂:雾状水、泡沫、二氧化碳、干粉。
第六部分:泄漏应急处理
应急处理:迅速撤离泄漏污染区人员至上风处,并进行隔离,严格限制出入。切断火源。建议应急处理人员戴自给正压式呼吸器,穿防静电工作服。尽可能切断泄漏源。合理通风,加速扩散。喷雾状水稀释、溶解。构筑围堤或挖坑收容产生的大量废水。如有可能,将漏出气用排风机送至空旷地方或装设适当喷头烧掉。也可以将漏气的容器移至空旷处,注意通风。漏气容器要妥善处理,修复、检验后再用。
第七部分:操作处置与储存
操作注意事项:密闭操作,全面通风。操作人员必须经过专门培训,严格遵守操作规程。远离火种、热源,工作场所严禁吸烟。使用防爆型的通风系统和设备。防止气体泄漏到工作场所空气中。避免与氧化剂接触。在传送过程中,钢瓶和容器必须接地和跨接,防止产生静电。搬运时轻装轻卸,防止钢瓶及附件破损。配备相应品种和数量的消防器材及泄漏应急处理设备。
储存注意事项:储存于阴凉、通风的库房。远离火种、热源。库温不宜超过30℃。应与氧化剂等分开存放,切忌混储。采用防爆型照明、通风设施。禁止使用易产生火花的机械设备和工具。储区应备有泄漏应急处理设备。
第八部分:接触控制/个体防护
职业接触限值
中国MAC(mg/m3):未制定标准
前苏联MAC(mg/m3):300
TLVTN:ACGIH窒息性气体
TLVWN:未制定标准
监测方法:
工程控制:生产过程密闭,全面通风。
呼吸系统防护:一般不需要特殊防护,但建议特殊情况下,佩戴自吸过滤式防毒面具(半面罩)。
眼睛防护:一般不需要特殊防护,高浓度接触时可戴安全防护眼镜。
身体防护:穿防静电工作服。
手防护:戴一般作业防护手套。
其他防护:工作现场严禁吸烟。避免长期反复接触。进入罐、限制性空间或其它高浓度区作业,须有人监护。
第九部分:理化特性
主要成分:纯品
外观与性状:无色无臭气体。
pH:
熔点(℃):-182.5
沸点(℃):-161.5
相对密度(水=1):0.42(-164℃)
相对蒸气密度(空气=1):0.55
饱和蒸气压(kPa):53.32(-168.8℃)
燃烧热(kJ/mol):889.5
临界温度(℃):-82.6
临界压力(MPa):4.59
辛醇/水分配系数的对数值:无资料
闪点(℃):-188
引燃温度(℃):538
爆炸上限%(V/V):15
爆炸下限%(V/V):5.3
溶解性:微溶于水,溶于醇、。
主要用途:用作燃料和用于炭黑、氢、乙炔、甲醛等的制造。
其它理化性质:
第十部分:稳定性和反应活性
稳定性:
禁配物:强氧化剂、氟、氯。
避免接触的条件:
聚合危害:
分解产物:
第十一部分:毒理学资料
急性毒性:LD50:无资料
LC50:无资料
亚急性和慢性毒性:
刺激性:
致敏性:
致突变性:
致畸性:
致癌性:
第十二部分:生态学资料
生态毒理毒性:
生物降解性:
非生物降解性:
生物富集或生物积累性:
其它有害作用:该物质对环境可能有危害,对鱼类和水体要给予特别注意。还应特别注意对地表水、土壤、大气和饮用水的污染。
第十三部分:废弃处置
废弃物性质:
废弃处置方法:处置前应参阅国家和地方有关法规。建议用焚烧法处置。
废弃注意事项:
第十四部分:运输信息
危险货物编号:21007
UN编号:1971
包装标志:
包装类别:O52
包装方法:钢质气瓶。
运输注意事项:采用刚瓶运输时必须戴好钢瓶上的安全帽。钢瓶一般平放,并应将瓶口朝同一方向,不可交叉;高度不得超过车辆的防护栏板,并用三角木垫卡牢,防止滚动。运输时运输车辆应配备相应品种和数量的消防器材。装运该物品的车辆排气管必须配备阻火装置,禁止使用易产生火花的机械设备和工具装卸。严禁与氧化剂等混装混运。夏季应早晚运输,防止日光曝晒。中途停留时应远离火种、热源。公路运输时要按规定路线行驶,勿在居民区和人口稠密区停留。铁路运输时要禁止溜放。
第十五部分:法规信息
法规信息化学危险物品安全管理条例(1987年2月17日发布),化学危险物品安全管理条例实施细则(化劳发[1992]677号),工作场所安全使用化学品规定([1996]劳部发423号)等法规,针对化学危险品的安全使用、生产、储存、运输、装卸等方面均作了相应规定;常用危险化学品的分类及标志(GB13690-92)将该物质划为第2.1类易燃气体。
牛奶厂主要检测方式有什么
在盆地沉积物埋藏后所经历的成岩过程中,会发生复杂的微生物、有机质、水、岩之间的相互作用过程。若烃类发生侵位,还涉及烃类参与的反应。传统上往往将它们单独地分别研究。流体-岩石相互作用研究力图将烃源岩、储集岩矿物和孔隙流体(油、气、水)及其中的微生物作为一个完整的地球化学系统来研究其相互作用,这就要求进行沉积学、水文地质学、同位素地球化学、微生物学等多学科交叉研究,将地质观察、实验模拟、计算机模拟结合在一起,解决一些单一学科的问题。下面介绍实验地球化学测试、实验室模拟、热力学理论计算等方面的研究方法。计算机软件模拟将专门分章讨论。
一、实验地球化学测试
沉积盆地流体-岩石相互作用研究需要对储层中油、气、水、岩进行全面的分析。所分析的项目及数量取决于研究的内容和目标,不能一概而论。
1.分析测试内容
岩石分析 岩石的矿物成分、化学组成和储层物性;碳酸盐胶结物的碳、氧、锶同位素组成;硫酸盐和硫化物的产状、矿物习性、硫同位素组成;粘土矿物的X射线衍射分析和氧同位素分析。
流体包裹体分析 流体包裹体包括液相和气相包裹体,液相又包括水相和烃类。均一化温度是各类流体包裹体常分析的内容,用以确定胶结物形成时期、油气注入时间。对于水相包裹体,需测定Na、K、Ca、Cl组成及盐度,用激光拉曼光谱测定溶解的CH4、H2S、CO2气体质量分数,H2S硫同位素和CO2的碳同位素。对烃类包裹体则可进行全烃色谱分析,以确定是否发生蚀变。
油田水分析 用毛细管等速电泳或高效液相色谱(HPLC)分析有机酸中甲酸、乙酸、丙酸、丁酸、苯甲酸等的浓度及总量。利用等离子发射光谱(ICP)分析微量元素K、Sr、Mn、Al、Fe、Zn、B、Li、Cs、Cd等。用钼-硅法分析其中二氧化硅的含量。用质谱仪分析碳、氢、氧、硫、锶、硼的同位素组成。
烃类分析 分析稠油或沥青的物性和族组成、气相色谱特征、生物标志物和硫同位素,并与正常原油对比,以研究其成因机制。分析伴生气的气体组分和碳、硫同位素。
2.分析测试技术
国内众多的实验室已建立起了成熟的方法,来分析上述岩石学、流体包裹体及烃类分析的项目。唯粘土矿物(高岭石、蒙脱石和伊利石)的氧同位素分析国内尚未开展,但国外已有报道。油田水有机组分、微量元素及同位素分析,尚未为人熟知,有必要简要介绍。
1)有机酸分析技术
(1)等速电泳法(ITP) 该法采用在中空的毛细管内进行恒流电泳的独特的分离分析方法。油水样经水相蒸发预处理,除去大量无机盐类后,即可直接进样进行有机酸分离。所用仪器为瑞典LKB-2127等速电泳仪及岛津IP-2A型等速电泳仪,检测器为电导检测器、紫外检测器及电位梯度检测器,配以200mm×0.5mm聚四氟乙烯毛细管(LKB-2127)及50cm×1mm、100cm×0.5mm两级聚四氟乙烯毛细管(IP-2A)。采用电解质溶液及尾随电解质溶液分别为组氨酸盐+组氨酸溶液及2-N吗啉代乙磺酸溶液,或为HCl+β-丙氨酸溶液及正己酸溶液。水相蒸发处理过程为:取水样低温蒸发,调至酸性,然后以丙酮洗涤过滤,再调节至碱性,浓缩定容。方法的回收率及相对标准偏差分别为96%~105%和2.4%~7.6%。
(2)区带电泳法(CZE) 由于油田水中Cl-干扰测定结果,等速电泳法需对样品进行水相蒸发预处理,采用区带电泳法则避免了上述预处理。所用仪器为惠普HP3PCE高效毛细管电泳仪,毛细管为50cm×50μm内径熔融石英毛细管(有效长度48.5cm),检测器为二极管阵列检测器。电解质体系为:①邻苯二甲酸氢钾+十六烷基三甲基溴化铵,pH=6.0;②3,5-二硝基苯甲酸+十六烷基三甲基溴化铵+5%甲醇,pH=9.0。检测波长为254nm及210nm,间接检测,压力样进,油田水样过滤后,即可直接进样进行有机酸分离。方法的相对标准偏差为1.1%~3.5%。
(3)毛细管气相色谱法(GC) 利用AT1000大口径极性毛细管柱,对油田水中C2—C5一元羧酸进行分离分析。对油田水以水相蒸发除去大量无机盐类后,经浓缩再直接进样,无需酸化和萃取。方法回收率和相对标准偏差分别为79.6%~100%及1.9%~6.4%。
2)同步辐射X射线荧光分析
利用北京正负电子对撞机国家实验室同步辐射装置,在专用模式下进行工作。实验测试时,样品受同步辐射X射线激发,发生电离,被电离的原子产生次级特征X射线。每种元素有其固有的特征X射线能量及相应的特征波长,用Si(Li)探测器测定这些特征X射线的能量可判断元素的类别;根据测得的待测元素的特征X射线荧光计数与相同实验条件下标样所测的该元素的计数比较,可得出元素的含量。
由于同步辐射具有高亮度、高准直、线偏振及宽频可调等优异特性,因而用于样品的微量元素分析时灵敏度高,对制样要求简单,可在保持样品原始状态下进行测定,并能在相同的实验条件下同时测定一个油田水样品中的20多种微量元素,检测下限可达10-6量级。
3)δD、δ18O、δ34S和87Sr/86Sr的测定
δD的测试采用的是高纯锌(Zn)还原法,即将2μL水样在390℃下经过锌还原出氢气,然后用MAT251质谱仪测定氢气的D/H值。δ18O的测量采用CO2-H2O平衡法,即将一定量的CO2高纯钢瓶二氧化碳与2mL水样平衡,用MAT251型质谱仪测定平衡后CO2的18O/16O。δD、δ18O测试结果均以SMOW(标准平均大洋水)为标准给出,其标准偏差分别为1‰~2‰和0.20‰~0.30‰。
δ34S硫化物硫同位素分析方法是,将硫化物与一定比例CuO混合,在1100℃下真空燃烧制备纯的SO2气体。硫酸盐、自然硫或岩石中微量硫,均采用埃斯卡试剂处理,转化为氧同位素基本纯的硫酸钡。制样时,称取一定量的BaSO4、V2O5、SiO2(比例为1∶3.5∶3.5),混合均匀后放入瓷瓶内,并在其上覆盖一层铜丝,在980℃的真空热解下,制备纯的SO2气,然后用MAT251型质谱计测定34S/32S值。δ34S值以CDT(为迪亚布洛峡谷陨石中的陨硫铁)标准给出。其标准偏差为±0.10‰~0.30‰。
87Sr/86Sr测定方法是,取一定量地层水,用超纯HCl酸化,经过标准离子交换技术分离后,在MTA261型多接收器质谱仪上进行测定。溶解碳酸盐全岩、胶结物是用超纯的HCl,溶解页岩采用超纯HF和HClO4试剂。分析精度0.00003~0.00007。其中,地层水样来自中途测试或完井测试。但是,这类样品不可能有足够的采样覆盖面,尤其在井内更是如此。最有效的弥补方法是使用岩心样品,这就涉及岩心的保护及其水的离心分离。在应用了低浸染取心技术(即最大限度地减少泥浆对水的污染)以后,这种方法非常实用。还有一种是RSA法,即残余盐分析法。在实验室中用超纯水浸滤未经保护的常规岩心,以溶解孔隙中的盐。这种盐是岩心在储藏期间从蒸发的地层水中沉淀出来的。由于不可能浸滤出100%的盐类物质,所以浸滤出的盐不保留原始地层水总体化学性质。但是通过对RSA法的有效性严格检验后,发现锶同位素87Sr/86Sr比值却不受影响。在取样过程中必须避免在岩心边缘、裂隙面和含有高渗透性岩石的部位取样,筛去具有污染特征的数据(取决于渗透性与87Sr/86Sr之间的关系),还要沿一些岩样的半径方向测定RSA法的数据特征,以此来校验岩心中央未被污染水的稳定比值。与多种钻井泥浆渗透液相比,地层水中的高Sr含量意味着水中87Sr/86Sr比对污染作用相对地不太敏感。比较而言,地层水的87Sr/86Sr比值为0.705~0.730,砂岩中矿物的87Sr/86Sr比值变化范围更大:斜长石或碳酸盐小于0.710,钾长石大于0.730,而云母大于0.800。可见,用RSA法可以将油田水87Sr/86Sr比值十分精确地测定出来(Smalley,1987)。
二、实验室模拟
模拟实验是在实验室中通过控制实验条件来模拟自然条件下流体-岩石相互作用的过程。模拟实验包括动力学和热力学两种模拟方法。中国地质科学院张荣华研究员一直在模拟研究开放体系中方解石、萤石等矿物-水的反应动力学。而沉积盆地水-岩反应更常发生在半封闭-半开放体系中。模拟的内容包括:有机酸、CO2的生成;有机组分(原油、有机酸等)参与的水-岩相互作用;金属有机配位化合物稳定性的实验测量等。常用的模拟实验方法是流动或动态实验装置(Barth等,1988;杨俊杰等,1995)。该方法是将反应溶液从一端注入,并在控制的温度、流速下与反应容器中涂有环氧树脂的岩心发生作用。反应溶液可以是各种合成地层水,可含有机酸或原油。在不同的持续时间里从另一端收集反应后的溶液,观测水化学的变化。另一方法采用间歇反应器(静态装置),反应容器可用不锈钢、钛制成。采集并分析经不同时间反应后的溶液,对比实验前后岩石的显微特征、物性或原油性质的变化,以达到模拟研究流体-岩石相互作用的目的。
三、热力学理论计算
热力学理论计算方法是运用热力学定律,对地球化学反应和过程进行理论计算来推断和解释各种地球化学现象(梅廉夫等,1994),可为实验结果的延拓、解释和检验提供理论依据。倪师军等(1993)根据流体包裹体温度、压力、成分及Eh-pH值,计算了成岩流体与矿物相互作用的趋势。而自由能更广泛应用于化学反应趋势的预测上。McBride(1987)、罗明高(1995)以反应的自由能模拟计算了成岩作用的序列;Meshri(1990)对比研究了碳酸和有机酸的热力学反应能力,计算了碳酸盐矿物方解石和铝硅酸盐矿物长石的溶解趋势和向粘土矿物转化趋势。Giles(1990)利用质量传递方程研究了矿物溶解-沉淀、离子迁移能力对次生孔隙和总孔隙度变化的影响。可见,热力学理论计算已用于地质现象的解释和预测上,是计算机软件模拟的基础。但相对而言,考虑的因素较为单一。
环状亚胺液相色谱方法
理化检测法: 理化检测法是利用抗生素分子中的基团所具有的特殊反应或性质来测定其含量的方法,如高效液相色谱
法、气相色谱法、比色法、荧光分光光度法等,最常用的是高效液相色谱及其联用技术。这类方法大多既能进行定性分析,也能进行定量检测,而且检测速度快,灵敏度、特异性和分辨率均很高,重复性好,应用广泛。但是其检测要求高,需要昂贵的检测设备,检测程序较复杂,检测费用较高,一般应用于科研,很难在基层推广应用。
1.1 高效液相色谱法
高效液相色谱法(HPLC)是一种快速的分离分析技术,其中反相HPLC发展最快。它引入了气相色谱理论,在技术上采用高压泵、高效固定相和高灵敏度检测器,实现了分离速度快、效果好和操作自动化,几乎所有的化合物包括高极性/离子型待测物和大分子物质均可用HPLC进行测定。其原理是利用液体作为流动相,通过高压,使被测样品和流动相经过色谱柱,样品中的成分在色谱柱中反复分配,使各组分分离,然后由检测器检测各组分含量。由于HPLC不需高温条件,故适于易受热分解、不易气化的残留成分分析。HPLC的特点是灵敏度高、检测限低、方法稳定,因此广泛应用于青霉素类、四环素类等抗生素及呋喃类(呋喃西林、呋喃唑酮)药物、磺胺类药物的残留检测。但在利用高效液相色谱法检测牛奶中青霉素残留时,要经过样品处理(包括样品的提取、脱蛋白、离心、层析柱净化、衍生化等步骤)、残留药物分离和残留药物检测3步程序[3]Marchetti[4]等使用此方法检测牛奶中的青霉素G、青霉素V、苯唑西林、邻氯青霉素、双氯青霉素,其中青霉素G的检测限度为4μg/L,其它4种青霉素的检测限为10μg/L。Furusawa[5]使用HPLC法检测牛奶中青霉素G的残留量,检测灵敏度为0.004μg/mL。
1.2 色谱质谱联用技术
色谱质谱联用法可扬长避短,一般兼分离、定量和定性(分子结构信息)于一体,因而特别适用于确证性分析。联用技术实现了高效层析分离和检测联机,可用微电脑控制层析条件、程序并且可以进行数据处理,其特异性、灵敏度和重复性均较好,并可一次同时完成同一样本中多种药物及其代谢物检测。对于分析牛奶中青霉素类抗生素,质谱作为一种专一检测器已获得广泛应用。常见的联用技术有薄层色谱-质谱(TLC-MS)、气相色谱一质谱(GC-MS)、液相色谱-质谱(LC-MS)、超临界流体色谱-质谱(SFC-MS)等[6] 。
近年来,还有研究者尝试用高效薄层色谱(HPTLC)、超临界流体色谱(SFC)和毛细管区域电泳法(CZE)等进行抗生素残留的检测[7]。这些方法能对抗生素进行定性、定量测定,灵敏度较高,但一般需要昂贵的仪器设备、大量复杂的前处理、熟练的技术人员及较长的分析周期,而且只能用于单个样本的检测,因而不常用。
2 微生物学检测方法
目前,牛奶中抗菌药物残留的微生物学分析方法一般分为微生物生长抑制法、微生物受体法和酶促比色法。
2.1 微生物生长抑制法
其测定原理基于抗生素对微生物的生理机能、代谢的抑制作用,可定性或定量确定样品中抗生素的残留。其工作菌株大多选用枯草芽孢杆菌、嗜热脂肪芽孢杆菌、藤黄八叠球菌等敏感菌株[8]。目前,常用的微生物生长抑制法主要有以下几种。
2.1.1 纸片法
即PD法(Paper Disc)[9],当样品中存在抑菌物质时,在纸片周围形成一个清晰的抑菌圈。在检测过程中,培养基内加入溴甲酚紫指示剂,当样品中含有抑菌物质时,纸片周围有一个清晰的浅蓝色的抑菌圈。抑菌圈的大小决定于抑菌物质的种类和浓度。检测限可达0.008IU/mL以下,一般在4h内可获得结果。
2.1.2 戴尔沃-p法和戴尔沃-p多次测试法[10]
戴尔沃-p法是将牛奶样品和含有营养剂及pH值指示剂的片剂加入到安瓿瓶中。安瓿瓶中已含有接种了嗜热脂肪芽孢杆菌的琼脂,当没有抑菌物质时,细菌生长产生足量的酸,使内含的pH值指示剂发生颜色变化,从紫色变为蓝色;当存在抑菌物质时,细菌的正常生长被抑制,产酸被延滞,内含的pH值指示剂不发生颜色变化。戴尔沃-p多次测试法是Delvotest-p法的改良方法,它们的原理一样,但戴尔沃-p多次测试法不使用安瓿瓶,而是使用具有96个测试孔的聚苯乙烯板,优点是一次能做96个试验。随着检测方法的发展,这2种方法已被美国官方分析师协会(AOAC)所接受,用于乳和乳制品中β-内酰胺类抗生素的检测。
2.1.3 亮黑还原法[11]
简称BR-test。测试时,在样品中加入含有嗜热脂肪链球菌的琼脂片,然后培养。在培养期间,菌种繁殖使内含的氧化还原指示剂还原成**。当样品中含有抑菌物质时,菌种生长受抑制,培养基琼脂保持亮黑指示剂原来的蓝色。BR-test法已改良过多次,其中有名的BR-test“Blue Star”法,已被加拿大官方接受使用[12]。
除了这些检测方法外,藤黄微球菌法( Sarcina lutea test)、三磷酸腺苷法(ATP test)、三碟法( three-plate test)、六碟法( six-plate test) 等都是微生物抑制法。这些方法无一例外地都需要对微生物进行培养,同时存在检测时间长,检测极限难以满足要求,人为因素影响大及很难实现标准化定量检测等缺点。但由于成本较低,在我国基层乳品企业还有应用。
2.2 微生物受体法
微生物受体分析法CHARM I和CHARM II比生长抑制分析法用途更广,可适用于检测不同基质样品中各种抗生素残留。CHARM I是专用于牛奶中β-内酰胺抗生素残留的检测方法,也是美国AOAC承认的第一个快速测定法。这个方法非常快速和灵敏,一个牛奶样品分析只需15min。CHARM II法的原理是,一类抗生素的功能团和加入的微生物细胞壁上或细壁内的受体位点进行不可逆的结合反应。该法往往利用同位素标记的抗生素与样品中的抗生素残留竞争结合位点,当样品中含有抗生素残留时,残留的抗生素分子阻止放射性标记的抗生素与受体位点结合。因此,在位点上结合的标记物越多,说明样品中抗生素越少。在基质中添加一定浓度的抗生素来建立一个控制点,用于判断样品是否存在抗生素残留。
2.3 酶比色法[13]
酶比色法来自于Penzyme法,是用于快速检测牛奶中β-内酰胺类抗生素的定量酶比色法。其原理是测定β-内酰胺类抗生素对DD-羧肽酶灭活的程度。在此酶的存在下,链酶菌会释放D-丙氨酸,D-丙氨酸被氧化成丙酮酸的同时会产生过氧化氢,而过氧化氢可使用氧化还原显色指示剂来测定。检测时间为15min,指示剂**不变,则结果为阳性;如指示剂变为粉红色,则结果为阴性;颜色处于粉红和**中间,则牛奶中的β-内酰胺类抗生素的量可以定量估计在0~0.017IU/mL之间。
3 免疫分析法
目前,应用的抗生素残留免疫分析技术主要分为两大类:一是以抗原抗体识别为核心反应的酶联免疫吸附法;二是以受体配体识别为核心反应的受体吸附分析法。
3.1 酶联免疫吸附法
ELISA是当前应用最广、发展最快的免疫测定技术,具有灵敏度高,特异性强,处理量大等特点。当前关于ELISA法检测抗生素残留的研究报道以竞争ELISA法为主[14]。Strasser[15]等采用直接竞争ELISA法检测牛奶中青霉素类抗生素残留,检测范围为2~32ng/mL;Samsonova[16]等用间接竞争ELISA法检测牛奶中氨苄青霉素残留,最小检测限为5.0ng/mL。Cliquet[17]等利用间接竞争ELISA法可同时检测到氨苄青霉素、青霉素G、羟氨青霉素、苯唑青霉素、双氯青霉素,检测灵敏度都在欧盟最大残留量(MRL)限度内。
3.2 受体吸附分析法
受体吸附分析法是继免疫吸附法之后检测抗生素残留的受体配体之间的特异性识别反应。国外在此领域研究较早,我国的研究由于起步较晚,目前未见相关报道[18]。Setford[19]等应用固定了青霉素结合蛋白的工作电极检测牛乳中青霉素G残留,当样品中青霉素G浓度分别为5μg/kg和10μg/kg时,测得的抑制率分别为33.5%和77.1%。此方法的变异系数为4.2%~26.4%。近年来免疫检测又出现了许多新技术,如免疫传感器、流动注射免疫分析技术、荧光偏振光免疫分析等。Eva Gustavsson[20]等使用表面等离子共振传感器(SPR),设计了具有羧肽酶活性的受体蛋白活性抑制试验以检测牛乳中β-内酰胺残留。通过相应抗体来检测底物量进而计算羧肽酶活力的变化,实现了定量检测牛乳中的青霉素G残留。此方法对于青霉素G的检测极限为2.6μg/kg;连续3天测定已知青霉素G残留量为4μg/kg的样品,此方法的变异系数为7.3%~16.0%,回收率为108%~118%。Cacciatore[21]等使用等离子共振传感器检测了牛乳中β-内酰胺抗生素残留。其反应通过地高辛标记的氨苄青霉素与样品中β-内酰胺抗生素竞争固定于等离子共振传感器传感片表面的青霉素受体的结合位点,实现了牛乳中β-内酰胺抗生素的定量检测。此方法对青霉素G的检测极限是4μg/kg 。
东部X市浅层地下水有机污染现状及其安全性评价
高效液相色谱法按分离机制的不同分为液固吸附色谱法、液液分配色谱法(正相与反相)、离子交换色谱法、离子对色谱法及分子排阻色谱法。 1.液固色谱法 使用固体吸附剂,被分离组分在色谱柱上分离原理是根据固定相对组分吸附力大小不同而分离。分离过程是一个吸附-解吸附的平衡过程。常用的吸附剂为硅胶或氧化铝,粒度5~10μm。适用于分离分子量200~1000的组分,大多数用于非离子型化合物,离子型化合物易产生拖尾。常用于分离同分异构体。 2.液液色谱法 使用将特定的液态物质涂于担体表面,或化学键合于担体表面而形成的固定相,分离原理是根据被分离的组分在流动相和固定相中溶解度不同而分离。分离过程是一个分配平衡过程。 涂布式固定相应具有良好的惰性;流动相必须预先用固定相饱和,以减少固定相从担体表面流失;温度的变化和不同批号流动相的区别常引起柱子的变化;另外在流动相中存在的固定相也使样品的分离和收集复杂化。由于涂布式固定相很难避免固定液流失,现在已很少采用。现在多采用的是化学键合固定相,如C18、C8、氨基柱、氰基柱和苯基柱。 液液色谱法按固定相和流动相的极性不同可分为正相色谱法(NPC)和反相色谱法(RPC)。 正相色谱法 采用极性固定相(如聚乙二醇、氨基与腈基键合相);流动相为相对非极性的疏水性溶剂(烷烃类如正已烷、环已烷),常加入乙醇、异丙醇、四氢呋喃、三氯甲烷等以调节组分的保留时间。常用于分离中等极性和极性较强的化合物(如酚类、胺类、羰基类及氨基酸类等)。 反相色谱法 一般用非极性固定相(如C18、C8);流动相为水或缓冲液,常加入甲醇、乙腈、异丙醇、丙酮、四氢呋喃等与水互溶的有机溶剂以调节保留时间。适用于分离非极性和极性较弱的化合物。RPC在现代液相色谱中应用最为广泛,据统计,它占整个HPLC应用的80%左右。 随着柱填料的快速发展,反相色谱法的应用范围逐渐扩大,现已应用于某些无机样品或易解离样品的分析。为控制样品在分析过程的解离,常用缓冲液控制流动相的pH值。但需要注意的是,C18和C8使用的pH值通常为2.5~7.5(2~8),太高的pH值会使硅胶溶解,太低的pH值会使键合的烷基脱落。有报告新商品柱可在pH 1.5~10范围操作。 正相色谱法与反相色谱法比较表 正相色谱法 反相色谱法 固定相极性 高~中 中~低 流动相极性 低~中 中~高 组分洗脱次序 极性小先洗出 极性大先洗出 从上表可看出,当极性为中等时正相色谱法与反相色谱法没有明显的界线(如氨基键合固定相)。 3.离子交换色谱法 固定相是离子交换树脂,常用苯乙烯与二乙烯交联形成的聚合物骨架,在表面未端芳环上接上羧基、磺酸基(称阳离子交换树脂)或季氨基(阴离子交换树脂)。被分离组分在色谱柱上分离原理是树脂上可电离离子与流动相中具有相同电荷的离子及被测组分的离子进行可逆交换,根据各离子与离子交换基团具有不同的电荷吸引力而分离。 缓冲液常用作离子交换色谱的流动相。被分离组分在离子交换柱中的保留时间除跟组分离子与树脂上的离子交换基团作用强弱有关外,它还受流动相的pH值和离子强度影响。pH值可改变化合物的解离程度,进而影响其与固定相的作用。流动相的盐浓度大,则离子强度高,不利于样品的解离,导致样品较快流出。 离子交换色谱法主要用于分析有机酸、氨基酸、多肽及核酸。 4.离子对色谱法 又称偶离子色谱法,是液液色谱法的分支。它是根据被测组分离子与离子对试剂离子形成中性的离子对化合物后,在非极性固定相中溶解度增大,从而使其分离效果改善。主要用于分析离子强度大的酸碱物质。 分析碱性物质常用的离子对试剂为烷基磺酸盐,如戊烷磺酸钠、辛烷磺酸钠等。另外高氯酸、三氟乙酸也可与多种碱性样品形成很强的离子对。 分析酸性物质常用四丁基季铵盐,如四丁基溴化铵、四丁基铵磷酸盐。 离子对色谱法常用ODS柱(即C18),流动相为甲醇-水或乙腈-水,水中加入3~10 mmol/L的离子对试剂,在一定的pH值范围内进行分离。被测组分保时间与离子对性质、浓度、流动相组成及其pH值、离子强度有关。 5.排阻色谱法 固定相是有一定孔径的多孔性填料,流动相是可以溶解样品的溶剂。小分子量的化合物可以进入孔中,滞留时间长;大分子量的化合物不能进入孔中,直接随流动相流出。它利用分子筛对分子量大小不同的各组分排阻能力的差异而完成分离。常用于分离高分子化合物,如组织提取物、多肽、蛋白质、核酸等。 色谱法的基本原理 利用样品混合物中各组分理、化性质的差异,各组分程度不同的分配到互不相溶的两相中。当两相相对运动时,各组分在两相中反复多次重新分配,结果使混合物得到分离。 两相中,固定不动的一相称固定相;移动的一相称流动相。 分类: 根据流动相分—以气体作流动相—气相色谱——固定相为液体 气-液色谱 固定相为固体 气-固色谱 —以液体作流动相—液相色谱——固定相为液体 液-液色谱 固定相为固体 液-固色谱 —当流动相是在接近它的临界温度和压力下工作的液体时——超临界色谱 根据固定相的附着方式 —固定相装在圆柱管中—柱色谱 —固定相涂敷在玻璃或金属板上—薄膜色谱(平板色谱) —液体固定相涂在纸上—纸色谱(平板色谱) 根据分离机理 —分配色谱—样品组分的分配系数不同 —吸附色谱— 样品组分对固定相表面吸附力不同 —体积排阻色谱—利用固定相孔径不同,把样品组分按分子大小分开 —离子交换色谱—不同离子与固定相商相反电荷间的作用力大小不同 根据极性 —流动相极性>固定相极性-反相色谱 —流动相极性<固定相极性-正相色谱 气相色谱只适合分析较易挥发、且化学性质稳定的有机化合物,而HPLC则适合于分析那些用气相色谱难以分析的物质,如挥发性差、极性强、具有生物活性、热稳定性差的物质。所以,HPLC的应用范围已经远远超过气相色谱。 一、吸附色谱(adsorption chromatography) 又叫液固色谱法:流动相是液体,固定相是固体。 分离原理:固定相是固体吸附剂,吸附剂是多孔性微粒物质表面有吸附中心。样品组分与流动相竞争吸附中 心。各组分的吸附能力不同,使组分在固定相中产生保留时间不同和实现分离。 固定相: 固定相通常是强极性的硅胶、氧化铝、活性炭、聚乙烯、聚酰胺等固体吸附剂。活性硅胶最常用。 流动相: 弱极性有机溶剂或非极性溶剂与极性溶剂的混合物,如正构烷烃(己烷、戊烷、庚烷等)、二氯甲 烷/甲醇、乙酸乙酯/乙腈等。 应用: 对于极性,结构异构体分离和族分离仍是最有效的方法,如农药异构体分离、石油中烷、烯、芳烃的 分离。 缺点是容易产生不对称峰和拖尾现象。 二、分配色谱 原理: 固定液机械的吸附在惰性载体上,样品分子依据他们在流动相和固定相间的溶解度不同,分别进入两相分配而实现分离。 固定相:将一种极性或非极性固定液吸附在惰性固相载体上。如全多孔微粒硅胶吸附剂。 根据极性不同分类:正相分配色谱—固定相载体上涂布的是极性固定液; 流动相是非极性溶剂; 可分立极性较强的水溶性样品; 弱极性组分先洗脱出来。 反相分配色谱—固定相载体上涂布的是非极性或弱极性固定液; 流动相是极性溶剂; 强极性组分先洗脱出来。 液-液分配色谱固定相中的固定液体往往容易溶解到流动相中去,所以重现性很差,且不能进行梯度洗脱,已经不大为人们所采用。 三、键合相色谱 考虑分配色谱法中固定液的缺点,因此将各种不同的有机关能团通过化学反应共价结合到固定相惰性载体上,固定相就不会溶解到流动相中去了。 键合固定相优点:○ 对极性有机溶剂有良好的化学稳定性 ○使色谱柱的柱效高、寿命长 ○实验重现性好 ○几乎适于各种类相的有机化合物的分离,尤其是k’宽范围的样品 ○可以梯度洗脱 根据极性不同分类:正相键合相色谱—固定相极性>流动相极性 固定相:二醇基、醚基、氰基、氨基等极性基团的有机分子。 适于分离脂荣、水溶性的极性、强极性化合物 反相键合相色谱—固定相极性<流动相极性 固定相:烷基、苯基等非极性有机分子。如最常用的ODS柱或C18柱就 是最典型的代表,其极性很小。 适于分离非机性、弱极性离子型样品, 是当今液相色谱的最主要分离模式。 正相HPLC(normal phase HPLC): 是由极性固定相和非极性(或弱极性)流动相所组成的HPLC体系。其代表性的固定相是改性硅胶、氰基柱等,代表性的流动相是正己烷。吸附色谱也属正相HPLC。 反相HPLC(reversed phase HPLC): 由非极性固定相和极性流动相所组成的液相色谱体系,与正相HPLC体系正好相反。其代表性的固定相是十八烷基键合硅胶(ODS柱,Octa Decyltrichloro Silane),代表性的流动相是甲醇和乙腈。 四、体积排阻色谱(SEC,size exclusion chromatograghy) (又称凝胶色谱和分子筛色谱) 原理: 以多孔凝胶(如葡萄糖,琼脂糖,硅胶,聚丙烯酰胺等)作固定相,依据样品分子量大小达到分离目 的。大分子不进入凝胶孔洞,沿多孔凝胶胶粒间隙流出,先被洗脱;小分子进入大部分凝胶孔洞, 在柱中被强滞留,后被洗脱。 根据样品性质分类:凝胶过滤(GFC)—用于分析水溶性样品,如多肽、蛋白、生物酶、寡聚核苷酸、多聚核 苷酸、多糖。 凝胶渗透(GPC)—用于分析脂溶性样品,如测定高聚物的分子量。 SEC主要依据分子量大小进行分离,因此与样品、流动相间的相互作用无关。因此不采用改变流动相的组成来改善分离度。 五、离子交换色谱 (ion exchange chromatography, IEC) 分离原理:使用表面有离子交换基团的离子交换剂作为固定相。带负电荷的交换基团(如磺酸基和羧酸基)可以用于阳离子的分离;带正电荷的交换基团(如季胺盐)可以用于阴离子的分离。不同离子与交换基的作用力大小不同,在树脂中的保留时间长短不同,从而被相互分离
醇能跟钠反应吗,产物有哪些,会不会产生氢气
俞光明1 刘红樱2 苏晶文2 张泰丽2 黎伟1 沈莽庭2
(1.浙江省地质环境监测总站,杭州310007;2.南京地质矿产研究所,南京210016)
摘要:针对东部X市的浅层地下水样品进行了有机污染物的定量分析,研究了浅层地下水中有机污染物的分布特征,并参照检出限和饮用水标准值,采用污染指数法对浅层地下水的有机污染进行了初步评价,同时探讨了其来源。结果表明:X市浅层地下水中普遍检出的卤代烃是三氯甲烷,其次为三溴甲烷,多环芳烃是亚二氢苊、二氢苊、菲、蒽、芘、苯并[a]蒽、苯并[b]荧蒽、荧蒽、萘和屈,有机氯农药是p,p′-DDT、o,p′-DDT、γ-六六六、p,p′-DDD和δ-六六六。X市浅层地下水受到轻度的有机污染,其中环境影响度较大的P,P′-DDT和苯并[a]蒽应引起重视。根据地球化学参数和浅层地下水环境背景推测有机污染物分别来自于各种类型的工矿企业、化肥农药的施用、垃圾填埋场和化石燃料的不完全燃烧,城市不同功能区存在差异。
关键词:浅层地下水;有机物污染;安全性评价;中国东部城市
X市是我国东部发达地区迅速崛起的一座城市,处江、海、湖、河交会之位,其人口稠密,水乡特色浓郁,工农业并举,其地下水开发利用程度高,开采地下水类型齐全、开采范围较大、开采量较高,特别是居民生活饮用水水源、农业和农村生态建设等与地下水关系密切。随着社会经济的发展,地下水的污染负荷不断加大,特别有机污染物的排放与日俱增,而其中一些往往难以降解,并具有生物积累性和“三致”作用或慢性毒性,因此地下水有机污染将威胁到区内的饮水安全、生态环境和人体健康。而浅层地下水更易受到污染的影响,因此针对X市的浅层地下水有机污染状况进行调查评价,将为其地下水资源保护和有机污染防治提供依据,也为东部其他城市开展地下水有机污染调查评价提供借鉴。
1 样品采取与分析
针对东部地区的X市,依据农业区、居民区、商业区、工业区和垃圾场等城市功能区布置了浅层地下水样品(表1)。
表1 X市浅层地下水样品的分布
在野外调查的基础上,选择具有代表性、资料翔实、易于取样操作、相对稳定、并尽可能与以往动态监测井重合的井点。
准备采样器材:顶空瓶、密封性好的聚四氟乙烯或玻璃容器、聚四氟乙烯封口膜。非挥发性有机物水样瓶用1:1硝酸浸泡过夜,自来水洗3次,去除有机物的水洗3次,丙酮洗2次,360℃下干燥1小时。
采样前清除井管的滞水,用所要采集的水冲洗采样器或采样瓶3~4次,然后将水样沿瓶壁缓缓加入瓶内,装样过程中,瓶内气泡不得上下翻滚,使水样尽量充满。装满采样瓶后,封口钳封盖或瓶口用聚四氟乙烯的膜盖上,然后塞紧盖子,翻转瓶子检查,如存有气泡,重新取样。样品避光低温保存,有条件的4℃下保存或用冰块保存,并隔离放置。在取样中选取10%的样品采集密码平行样,并按5%的比例采集野外空白样品。
分析浅层地下水样品7件。分析依据方法:水质,挥发性卤代烃的测定,气相色谱法,GB/T17130-1997;水质,苯系物的测定,气相色谱法,GB/T11890-1989;水质,六六六、滴滴涕的测定,气相色谱法,GB/T7492-1987;16种多环芳烃,USEPA方法525.2饮用水中有机化合物的测定,液相萃取、毛细管拄气相色谱/质谱法。样品分析测试由中国地质大学(北京)地学实验中心承担,仪器设备为HPGC-6890气相色谱仪,编号:19990006。测试过程质量监控。
共检测浅层地下水中有机物44项,其中卤代烃7项,苯系物(单环芳烃)7项,多环芳烃16项,有机氯农药14项。
2 评价方法
2.1 浅层地下水有机污染评价方法
测试的45种浅层地下水中有机化合物为毒理学指标,采用地下水中i组分的实测浓度Ci与其检出限(对照值C0max)和饮用水标准值 (表1)对比进行浅层地下水有机污染评价。
未污染:Ci≤检出限;轻度污染:检出限<Ci≤1/2×饮用水标准值;中度污染:1/2×饮用水标准值<Ci≤饮用水标准值;重度污染:饮用水标准值<Ci≤5×饮用水标准值;极度污染:Ci>5×饮用水标准值。
2.2 浅层地下水有机物安全性评价方法
浅层地下水中某一有机物的安全性采用环境影响度AS(Ambient Severit)评价方法,即:
华东地区地质调查成果论文集:1999~2005
式(1)中:ASi为某种有机物在浅层地下水中的环境影响度;Ci为某种有机物在浅层地下水中的浓度;
华东地区地质调查成果论文集:1999~2005
为某种有机物i在水体中的目标值,根据水环境目标值华东地区地质调查成果论文集:1999~2005
、WHO推荐值[4]、国标值[1]等的最低值确定。AMEG即周围多介质环境目标值(Ambiet Multimedia EnvironmentalGoals),是美国环境保护局工业环境实验室推算出来的化学物质或其降解产物在环境介质中的限定值[3]。某一浅层地下水体中有机物的环境安全性评价采用总环境影响度评价法。假定所有有机物只要AS相同,则对人和环境的潜在危害程度相同,同时假定AS值大小与潜在危害呈线性关系[3],则某浅层地下水体的环境危害程度可以近似地用各组分的AS之和TAS(称为总环境影响度)表示,即:
华东地区地质调查成果论文集:1999~2005
当TAS大于l时,表明浅层地下水体中有毒化合物对人体健康具潜在危害,水体中TAS值小于1则其潜在危害作用不明显。
3 结果与讨论
3.1 浅层地下水中有机物的检出情况
X市浅层地下水中主要检出的卤代烃是三氯甲烷,其次为三溴甲烷,高值点主要分布在农业区和垃圾场(图1,表2)。
图1 东部X市浅层地下水中有机物的检出情况
表2 东部X市浅层地下水有机物含量及初步评价结果(单位ng/L)
续表
X市浅层地下水中未检出单环芳烃。
亚二氢苊、二氢苊、菲、蒽、芘、苯并(a)荧蒽和苯并(b)荧蒽所有样品均检出,荧蒽检出率86%,萘和屈57%检出。高值点主要分布在农业区、商业区和居民区。芴、苯并[k]荧蒽、苯并[a]芘、二苯并[a,h]蒽、茚并[1,2,3-c,d]芘和苯并[g,h,i]苝均未检出。
有机氯农药检出率较高的是p,p′-DDT(86%),次为o,p′-DDT、γ-六六六(57%)和p,p′-DDD、δ-六六六(43%)。高值点主要分布在垃圾场和工业区,其次为农业区和居民区。
3.2 浅层地下水有机污染初步评价
初步评价表明X市浅层地下水受到了轻度的有机污染,主要有机污染物为多环芳烃、滴滴涕和六六六(表2)。
3.3 浅层地下水有机物安全性评价
X市浅层地下水中有机物最大环境影响度AS均小于或远小于1,表明目前对环境和人体健康危害不显著。AS较大的为P,P′-DDT(0.32)和苯并[a]蒽(0.18),其次是芘(0.042)、O,P-DDT(0.037)、P,P′-DDD(0.031)、屈(0.019)、δ-六六六(0.013)和苯并[b]荧蒽(0.011),其中前两者应引起重视。
X市浅层地下水中总环境影响度为0.07~0.43,平均0.20,其中以农业区和垃圾场的为高,各代表性城市功能区浅层地下水有机污染未显示出明显的潜在危害作用。
3.4 浅层地下水中有机物来源探讨
在地球化学和环境地球化学研究中,经常使用一些特征化合物指数并结合地区环境特点和工业布局、污染排放类型等来判断PAHs的来源。
表3 东部X市浅层地下水有机物安全性评价
续表
注:A.中国GB3838-2002集中式生活饮用水地表水源地特定项目标准限值[1];B.1998WHO饮用水质量基准推荐值[4];C.2002U.S.EPA现行饮用水标准[2];D.中国CJ/T206-2005城市供水水质标准[5];E.AMEGwh水环境目标值[3]。
4环及其以上的具有高分子量的PAHs主要来源于化石燃料高温燃烧,而低分子量(2~3环)的则来源于石油类污染[6]。炼焦、石油裂解、原油泄漏、煤焦油提炼、染料工业等废水排放和泄漏来源的PAHs菲/蒽比值(P/A)为4.4~7.9,而家庭及生活炉灶、工业锅炉、露天燃烧及失火、机动车及内燃机等燃烧过程和大气沉降来源的PAHs其P/A值>10[7]。荧蒽/芘(Fl/Py)的比值为1.4代表煤型燃烧产物来源,比值为1左右代表木材燃烧来源,而比值小于1则显示来源于石油[8]。
浅层地下水中PAHs的含量总体是以以3环>5环>4环>2环>6环的顺序递减。P/A值0.68~8.52,Fl/Py值0~1.03,显示其来源于燃烧源和石油源的混合特征。其中农业区A1、居民区R1和垃圾场W1样品2+3环PAHs含量远大于4环及以上PAHs,Fl/Py值(0.01~0.62)小于1,P/A值0.68~2.23,以废水排放和石油类泄漏来源为主;农业区A2和商业区C1样品2+3环PAHs含量小于或约等于4环及以上PAHs,Fl/Py值1.23~3.45,P/A值0~0.25,其主要来自化石燃料的不完全燃烧,包括工业燃煤、生活供暖用煤和交通污染。工业区样品2+3环PAHs含量远大于4环及以上PAHs,P/A值(6.60~8.52)较大,Fl/Py值(0.31~1.03)变化较大,为混合来源。
表4 东部X市浅层地下水中PAHs特征指数
4 结论
X市浅层地下水中主要检出的卤代烃是三氯甲烷,其次为三溴甲烷,高值点主要分布在农业区和垃圾场。亚二氢苊、二氢苊、菲、蒽、芘、苯并(a)荧蒽、苯并(b)荧蒽、荧蒽、萘和屈等10种多环芳烃普遍检出,高值点主要分布在农业区、商业区和居民区。有机氯农药检出率较高的是p,p′-DDT、o,p′-DDT、γ-六六六、p,p′-DDD和δ-六六六,高值点主要分布在垃圾场和工业区,其次为农业区和居民区。
初步评价表明X市浅层地下水受到了轻度的有机污染,主要有机污染物为多环芳烃、滴滴涕和六六六。
X市浅层地下水中有机物最大环境影响度和总环境影响度均小于或远小于1,表明目前对环境和人体健康危害不显著,但AS较大的P,P′-DDT和苯并[a]蒽应引起重视。
根据地球化学参数和浅层地下水环境背景推测有机污染物分别来自于各种类型的工矿企业、化肥农药的施用、垃圾填埋场和化石燃料的不完全燃烧,城市不同功能区存在差异。
参考文献
[1]国家环境保护总局,国家质量监督检验检疫总局.地表水环境质量标准GB3838-2002.2002
[2] U.S. Environmental Protection Agency. 2002 Edition of the Drinking Water Standards and Health Advisories. 2002
[3] 汪晶,和德科,汪尧衢等.环境评价数据手册-有毒物质鉴定值(第1版).北京:化学工业出版社,1988,2~423
[4] WHO. Guidelines for drinking-water quality, 2nd ed. Addendum to health criteria and other supporting information. Geneva: World Health Organization, 1998,2:123~152
[5] 中华人民共和国建设部.城市供水水质标准CJ/T 206-2005.2005
[6] Erwin L. Biodiversity and life support impacts of landuse in LCA. Jouenal of Cleanr Production, 2000,8:313~319
[7] Gschwend P M,Hites R A. Fluxes of polycyclic aromatic hydrocarbons to marine and lacustrine sediments in the northeastern United States. Geochimica et cosmochimica Acta, 1981,45:2359~2367
[8] Edward N T. Polycyclic aromatic hydrocarbons (PAHs) in the terrestrial environment: A review. J. Environ. Qual. 1983, 12:427 ~432
[9] Simoneit B R T. Organic matter of the troposphere characterization and sources of petroleum and pyrogenic residues in acrosols over in western United States. Atoms. Environ.,1984.,18:51 ~67
Status of the Organic Pollution for the Surficial Groundwater in X City of Eastern China and Its Security Assessment
Yu Guangming1, Liu Hongying2, Su Jingwen2,Zhang Taili2,Li Wei1, Shen Mangting2
(1. Zhejiang General Monitoring Station for Geo-environment, Hangzhou 310007;2. Nanjing Institute of Geology and Mineral Resources, Nanjing 210016)
Abstract: Based on the concentration of organic pollutants in the surficial groundwater samples collected from X city of Eastern China were investigated and quantitatively analyzed, the distributing characteristics of these compounds were studied, the Pollution Index Mathod was adopted to assess the organic pollution of the surficial groundwater in X city by reference to minus detected limit and the drinking water Standard of China, WHO and U. S. EPA, and the sources of these compounds were discussed.
Results show that Trichloromethane, then Tribromomethane of chlorinated solvents, Acenaphthene, Acenaphthylene, Phenanthrene,Anthracene, Pyrene, Benzo [a]) acenaphthene, Benzo [b] fluoranthene, Fluoranthene, Naphthalene and Chrysene of PAHs, p, p′-DDT, o, p′-DDT, γ-BHC, p, p′-DDD and δ-BHC of organochlorine pesticides are mostly found in the surficial groundwater of X city. the surficial groundwater in X city mostly are polluted in light degree, in which p, p′-DDT and Benzo [a] Anthracene having bigger Ambient Severit (AS) should be considered. Depending on the geochemistry parameters of the organic pollutants and the environmental setting of the surficial groundwater, various types industrial production areas, use of fertilizer and pesticides, waste landfill and the incomplete pyrolysis of the fuel may be the various source of the organic pollutants in X city, there existed differences in various function district of city.
Key words: Surficial groundwater; Organic pollution; Security assessment; City in Eastern China
蒽醌的生产方法
醇羟基氢反应
由于醇羟基氢具定性醇金属钠反应氢氧键断裂形醇钠(CH3CH2ONa)放氢气
由于液相水酸性比醇强所醇与金属钠反应没水金属钠反应强烈若醇钠放入水醇钠全部水解醇氢氧化钠虽工业制甲醇钠或乙醇钠用醇与氢氧化钠反应设水除使平衡利于醇钠用利用形共沸混合物水带走转移平衡所沸共合物指几种沸点同完全互溶液体混合物由于间作用力蒸馏程气相液相组相同能具低沸点(比所组沸点都低)或高沸点(比所组沸点都高)馏物些馏物组与溶液组相同直蒸完沸点直恒定乙醇苯水组三元共沸混合物其沸点64.9℃(乙醇18. 5%苯74%水7.5%)苯乙醇组二元共沸混合物其沸点68.3℃(乙醇32.4%苯67. 6%)由于乙醇水形共沸混合物其沸点78℃(乙醇95. 57%水4. 43%)所乙醇含少量水能通蒸馏除计算加入比形乙醇苯水三元共沸混合物稍量苯先水除量苯与乙醇形二元共沸混合物除剩水乙醇醇钠醇溶液通述水醇钠及其类似物机合类重要试剂并作碱使用[2]
醇与含氧机酸反应
醇与含氧机酸反应失水机酸酯
醇与硝酸反应程:醇作亲核试剂进攻酸或其衍物带电荷部氮氧双键打醇氢氧键断裂硝酸部失水重新形氮氧双键
该类反应主要用于机酸级醇酯制备机酸三级醇酯制备宜用三级醇与机酸反应易发消除反应
醇与含氧机酸酰氯酸酐反应能机酸酯
含氧机酸酯许用途乙二醇二硝酸酯甘油三硝酸酯(俗称)都烈性炸药能用于血管舒张、治疗绞痛胆绞痛科家发现:能治疗脏病原能释放信使NO并阐明NO命作用机理荣获1998诺贝尔理医奖
命体核苷酸磷酸酯例甘油磷酸酯与钙离反应用控制体内钙离浓度反应失调导致佝偻病[2]
醇羟基取代反应
醇碳氧键极性共价键由于氧电负性于碳所其共用电偏向于氧亲核试剂进攻性碳碳氧键异裂羟基亲核试剂取代其重要亲核取代反应羟基卤原取代采用:
1.与氢卤酸反应
(1)般情况
氢卤酸与醇反应卤代烷反应醇羟基卤原取代
ROH+HX——>RX+H20
醇羟基离基团需要酸帮助使羟基质化水形式离各种醇反应性3°>2°>1°三级醇易反应需浓盐酸室温振荡即反应氢溴酸低温能与三级醇进行反应用氯化氢、溴化氢气体0℃通三级醇反应几钟内完制三级卤代烷用
氢卤酸氢碘酸酸性强氢溴酸其浓盐酸相弱卤离亲核能力I->Br->Cl-故氢卤酸反应性HI> HBr>HCl若用级醇别与三种氢卤酸反应氢碘酸直接反应氢溴酸需用硫酸增强酸性浓盐酸需与水氯化锌混合使用才能发反应氯化锌强路易斯酸反应作用与质酸类似
用Lucas试剂鉴别级醇、二级醇、三级醇
浓盐酸水氯化锌混合物称Lucas试剂用鉴别六碳六碳级、二级、三级醇别加入盛Lucas试剂试管经振荡发现三级醇立刻反应油状氯代烷溶于酸溶液呈混浊两层反应放热;二级醇2~5min反应放热明显溶液两层;级醇经室温放置1h仍反应必须加热才能反应
使用Lucas试剂须注意些级醇烯丙型醇(allylicalcohol)及苯甲型醇(benzylicalcohol)快发反应p-π共轭容易形碳离进行SN1反应
各类醇与Lucas试剂反应速率
烯丙型醇苯甲型醇三级醇>;二级醇>;级醇
氢卤酸与数级醇按SN2机理进行反应
氢卤酸与数二级、三级醇空阻特别级酵按SN1机理进行反应
按SN机理反应重排产物产2-戊醇与氢溴酸反应86%2-溴戊烷与14%3-溴戊烷;异丁醇氢溴酸与硫酸加热反应80%异丁基溴与20%三级丁基溴新戊醇由于β位位阻太重排产物2-甲基-2-溴丁烷三级醇与氢卤酸反应般发重排三级醇易发消除反应所取代反应需低温进行
2.与卤化磷反应
醇与卤化磷反应卤代烷
醇羟基离基团与三溴化磷作用形CH3CH2OPBr2Br进攻烷基碳原-OPBr2作离基团离- OPBr2两溴原继续与醇发反应
碘代烷由三碘化磷与醇制备通三碘化磷用红磷与碘代替醇、红磷碘放起加热先三碘化磷再与醇进行反应
氯代烷用五氯化磷与醇反应制备
述用三溴化磷与级醇、β位支链级醇、二级醇相应溴代烷用二级醇及些易发重排反应级醇温度须低于0℃避免重排红磷与碘用于级醇制相应碘代烷[2]
3.与亚硫酰氯反应
若用亚硫酰氯醇反应直接氯代烷同二氧化硫氯化氢两种气体反应程些气体都离反应体系利于反应向产物向进行该反应仅速率快反应条件温产率高且其副产物般用量亚硫酰氯并保持微沸制氧代烷[2]
4.经醇与磺酰氯反应间阶段制备卤代烃
醇羟基必须质酸或路易斯酸催化才进行取代反应苯磺酸酯酸根部离基团类酯比醇容易进行亲核取代反应
级或二级醇通与苯磺酰氯反应形磺酸酯再转卤代烷纯度磺酰氯由相应磺酸与五氯化磷反应制备[2]
醇氧化
级醇及二级醇与醇羟基相连碳原氢氧化醛、酮或酸;三级醇与醇羟基相连碳原没氢易氧化酸性条件易脱水烯碳碳键氧化断裂形化合物
1.用高锰酸钾或二氧化锰氧化
醇冷、稀、性高锰酸钾水溶液所氧化级醇、二级醇比较强烈条件(加热)氧化级醇羧酸钾盐溶于水并二氧化锰沉淀析羧酸
二级醇氧化酮由于二级醇用高锰酸钾氧化酮易进步氧化使碳碳键断裂故少用于合酮
三级醇性、碱性条件易高锰酸钾氧化酸性条件则能脱水烯再发碳碳键断裂化合物
高锰酸钾与硫酸锰碱性条件制二氧化锰新制二氧化锰β碳饱键级醇、二级醇氧化相应醛酮饱键受影响[2]
2.用铬酸氧化
铬酸作氧化剂形式:Na2Cr2O7与40%~50%硫酸混合液、CrO3冰醋酸溶液、CrO3与吡啶络合物等
级醇用NaCr2O7与40%~50%硫酸混合液氧化先醛醛进步氧化酸控制合适氧化条件氧化醛立即其反应体系蒸避免醛进步氧化酸反应需低于醇沸点高于醛沸点温度进行丙醇滴加温度~75℃NaCr2O7H2SO4H2O溶液旦丙醛蒸馏种反应产率高总部醛氧化酸醛沸点低于100℃才能用用途非限
二级醇用述几种铬酸氧化剂氧化酮条件比较稳定比较用
用铬酐(CrO3)与吡啶反应形铬酐双吡啶络合物吸潮性红色结晶称Sarrett(沙瑞特)试剂使级醇氧化醛二级醇氧化酮产率高吡啶碱性酸稳定醇种氧化剂反应般二氯甲烷于25℃左右进行双键、三键氧化受影响
二级醇Jones(琼斯)试剂氧化相应酮若反应物饱二级醇用Jones试剂氧化相应酮双键受影响该试剂铬酐溶于稀硫酸滴加要氧化醇丙酮溶液反应15~20℃进行较高产率酮
用量铬酸并反应条件强烈双键氧化酮或酸
用铬酐硫酸水溶液鉴别级醇、二级醇
级醇、二级醇使清澈铬酐硫酸水溶液由橙色变透明蓝绿色三级醇反应烯烃、炔烃反应述反应原级醇与二级醇起氧化作用[2]
3.用硝酸氧化
级醇能稀硝酸氧化酸二级醇、三级醇需较浓硝酸氧化同碳碳键断裂酸环醇氧化碳碳键断裂二元酸
4.Oppenauer氧化
另种选择性氧化醇叫做Oppenauer(欧芬脑尔)氧化(oxidation methods)即碱三级丁醇铝或异丙醇铝存二级醇丙酮(或甲乙酮、环酮)起反应(需加入苯或甲苯做溶剂)醇两氢原转移给丙酮醇变酮丙酮原异丙醇该反应特点醇酮间发氢原转移涉及其部所含碳碳双键或其酸稳定基团利用较适宜该由饱二级醇制备饱酮效[2]5.用Pfitzner—Moffatt试剂氧化
级醇Pfitzner(费兹纳)- Moffatt(莫发特)试剂作用产率非高醛试剂由二甲亚砜二环基碳二亚胺组二环基碳二亚胺英文名叫dicyclohexylcarbodiimide简称DCC二取代脲失水产物非重要失水剂(dehydrating agent)硝基苯甲醇磷酸试剂作用92%产率硝基苯甲醛
反应环基碳二亚胺接受水变脲衍物二甲亚砜变二甲硫醚氧化剂用于氧化二级醇
进行氧化反应必须注意:许机物与强氧化剂接触发强烈爆炸冈使用高锰酸钾、高氯酸及类似氧化剂定要溶剂进行反应溶剂使放量热消散减缓反应速率[2]
醇脱氢
级醇、二级醇脱氢试剂(dehydrogenating agent)作用失氢形羰基化合物醇脱氢般用于工业产用铜或铜铬氧化物等作脱氢剂300℃使醇蒸气通催化剂即醛或酮外Pd等作脱氢试剂[2]
3.4醇、酚醚卤素置换反应
3.4.1醇卤素置换反应
醇卤素置换反应获卤化物重要用卤化剂氢卤酸亚硫酰卤、磷酰卤及卤化磷等实际论哪种外乎先羟基变更离基团用卤素进行亲核取代
(1)氢卤酸(卤化氢)作卤化剂
醇与氢卤酸反应般亲核取代反应能形稳定碳离底物按SN1机理进行其反应通SN2机理
醇性顺序叔醇>仲醇>伯醇(SN1);氢卤酸(卤化氢)性顺序HI>HBr>HCl>HF低性卤化剂加入Lewis酸催化
叔胺催化类反应see RU 2051889, Process for Preparing 2-Ethylhexyl Chloride-1(1993).
(2)亚硫酰卤(卤化亚砜)作卤化剂
亚硫酰卤与醇反应卤代烷二氧化硫与卤化氢易离醇卤化应用较广孢哌酮钠间体氧哌嗪甲酰氯合
氯化亚砜氯化言同反应条件其反应机理尽相同
DMFHMPA催化醇与卤化亚砜反应DMF与SOCl2反应氯代烯铵盐:
该烯铵盐作氯化剂实现醇氯代
类似HMPA与SOCl2反应产物氯化剂其机理亦与DMF相同
反应示例:HMPA催化某伯醇氯代
机碱吡啶卤化氢盐提高卤离浓度能提高类反应速度该尤其适用于酸敏底物
取代或供电基取代芳醛与溴化亚砜共热二溴苄反应物存微量溴化氢醛羰基加反应第步
水DMF氯化亚砜芳醛转化相应二氯苄
(3)卤化磷作卤化剂
三卤化磷五卤化磷转化醇卤代烷用试剂其反应性较氢卤酸较少发重排反应用卤化磷三氯化磷三溴化磷者由溴素与磷原位制备
三卤化磷与醇反应亚磷酸单、双三酯卤离述磷酸酯发取代置换掉氧亚磷酰片断卤代烷
与卤化亚砜类似卤化磷与DMF反应能卤代烯铵盐者高性卤化剂与醇反应构型翻转卤代烷
(4)机膦卤化合物作卤化剂
三苯基膦卤化物Ph3PX2、Ph3PCX3X(PhO)3POX2等醇进行卤化反应性高条件温些卤化剂由三苯基膦或亚磷酸三苯酯与卤素或卤代烷原位合
其能反应机理其卤素取代程SN2反应
述反应DMF或HMPA进行使光性醇转化构型翻转卤代烷用于酸敏醇卤化
三苯基膦与N-卤代酰胺(NXS)反应产物类似机理发类似反应适用于酸稳定醇或甾醇卤化
(5)其卤化剂
卤硅烷试剂温条件醇转化卤代烷
NXS与二甲硫醚反应产物卤代硫鎓盐烯丙位苄位羟基取代高度选择性反应条件温影响其伯、仲羟基
甲磺酸/碘化钠温条件碘代烯丙位或苄位羟基种选择性能与碳离稳定性关
四甲基alpha-卤代烯胺温条件伯、仲羟基及烯丙位、炔丙位苄位羟基转化卤代烃其位阻类似物则高选择性卤代伯、烯丙位苄位羟基
其反应机理与DMF催化卤化亚砜卤化反应程相同
2-氯-3-乙基-苯并恶唑四氟硼酸盐类似反应机理温卤化剂前列腺素间体合
3.4.2酚卤素置换反应
酚羟基性低其卤置换反应必须使用五卤化磷或五卤化磷/氧卤化磷混合物较剧烈条件进行于某些底物单独使用氧卤化磷
三苯基膦卤化物转化酚芳卤化合物用试剂
例:
(R)-(+)- and (S)-(-)-2,2'-Bis(diphenyl phosphino)-1,1'-Binaphthyl(BINAP), Organic Syntheses, Coll. Vol. 8, P.57; Vol. 67, P.20.
羟基取代卤素形C-O-P结构反应第步继卤素CO取代(加消除)
3.4.3醚卤素置换反应
醚与氢卤酸等物质反应卤化物羟基化合物(醇或酚)四氢呋喃与甲醇氯化亚砜反应4-氯丁甲醚
醚氧原质化般反应第步亲核取代反应
醚键断哪片断醇哪片断卤化物取决于其吸电能力
BF3、BBr3等Lewis酸类似机理裂解醚键
see US 4595765, 1986.
三甲基硅卤化物温卤化剂
三甲基氯硅烷/碘化钠作碘化剂碘化氢高收率获碘化物
3.5羧酸卤素置换反应
见羧酸卤素置换反应羧羟基置换脱羧卤置换
3.5.1羧羟基卤素置换反应:酰卤制备
羧酸定条件与亚硫酰卤及卤化磷等卤化剂反应混酸酐-酰卤
(1)卤化亚砜作卤化剂
卤化亚砜较用羧酸卤化剂其优点于卤化卤化氢二氧化硫其本身沸点低、易除所反应易离
反应底物双键、羰基或酯基等影响较量卤化亚砜进行苯或石油醚等作溶剂
氯化亚砜由羧酸合酰氯用氯化剂与酸酐反应酰卤
反应机理:
反应机碱(吡啶或DMAP等)Lewis酸(ZnCl2等)催化
(2)卤化磷作卤化剂
卤化磷卤化剂性顺序五卤化磷>三卤化磷>三卤氧磷五氯化磷用于性较羧酸尤其具吸电基芳酸或芳香元酸酰氯化
产物应与三氯氧磷定沸点差利离
三卤化磷用于脂肪酸酰卤化
三氯氧磷与羧酸盐反应酰氯
反应机理三卤化磷例
述酰卤化反应羧酸性顺序脂肪酸>芳香酸(供电基取代芳酸>未取代芳酸>吸电基取代芳酸)说明羧羟基硫(磷)亲核进攻控制步骤
(3)草酰氯作氯化剂
草酰氯烃类溶剂温条件羧酸转化酰氯避免氯化磷等其氯化剂底物敏基团影响
反应机理:
见例转化羧酸钠盐避免氯化氢减少敏基团影响
(4)其卤化剂
氰脲酰氯(三聚氯氰)三乙胺存温条件羧酸转化酰氯
与醇卤化类似三苯基膦卤化物四甲基alpha-卤代烯胺用于由羧酸制酰卤反应
3.5.2羧酸脱羧卤素置换反应
羧酸银盐与溴或碘反应比底物少碳卤代烃称Hunsdiecker反应
于2-18碳饱脂肪酸言该反应般获较结同该反应用于芳香酸脱羧卤化
述反应若水存则影响收率甚至导致失败用汞盐代替稳定水银盐光照条件其收率高于银盐实际实施用羧酸、量氧化汞与卤素直接反应操作简单
与Hunsdiecker反应相似羧酸与金属卤化物(LiCl)、四乙酸铅苯或等溶剂反应脱羧氯化产物称Kochi改良
反应程发重排尤其适用于仲、叔氯代烃及beta-季碳氯代烃合
程能其反应机理
羧酸与碘、四乙酸铅四氯化碳进行光照发脱羧碘化反应称Barton改良伯或仲脂肪酸反应般较苯甲酸反应收率般60%左右
3.6其官能团卤素置换反应
3.6.1卤化物卤素交换反应
伯卤化物与机卤化物间卤素交换反应称Finkelstein反应
脂肪族卤化物反应应SN2机理
类似反应发芳香族卤化物
其反应机理能加-消除溶剂使用DMF、丙酮或二硫化碳等非质极性溶剂
Lewis酸通帮助卤素离化卤代烃所加入Lewis酸往往促进卤素交换反应
氟化用氟化钠、氟化钾、氟化银氟化剃等其氟化钾性较高、价廉用
氟化锑选择性与同碳原卤素反应与单卤素反应特性用合三氟甲基化合物
18-冠-6醚显著提高用氟化钾进行氟交换收率
3.6.2磺酸酯卤素置换反应
醇羟基转化性较高磺酸酯温条件卤代即避免醇卤化副反应比卤素交换效用卤化剂卤化钠、卤化钾、卤化镁卤化锂等
饱碳磺酸酯-卤素置换反应应SN2机理饱碳磺酸酯-卤素置换反应应加消除-机理
3.6.3芳香重氮盐卤素置换反应
芳香族重氮化合物卤素置换反应往往卤素引入直接卤化难引入位置反应卤化亚铜催化剂相应氢卤酸卤化剂进行称Sandmeyer反应
反应认自由基机理
芳香重氮盐碘置换反应加铜催化剂需重氮盐与碘素直接加热即
醇能跟钠反应吗,产物有哪些,会不会产生氢气
感觉提问主意不是很清晰
汽油分子浓度的检测?
精蒽氧化法气相固定床氧化法: 将精蒽加入气化室加热气化后与空气混合,二者比例为1︰(50~100)。混合气体进入氧化室,在V2O5催化下于(389±2) ℃下氧化,经薄壁冷凝后即得产品。
液相氧化法: 将精蒽计量后加入反应釜,再加入三氯苯在搅拌下溶解。然后滴加硝酸,控制反应温度105~110 ℃,将副产物NO排除,反应6~8 h后,减压蒸出溶剂,冷却结晶。得产品。此法设备腐蚀严重。苯酐法 将苯酐计量后加入反应釜,加苯在搅拌下加热熔解。加热至370~470 ℃,使混合气通过硅铝催化剂进行气相缩合。得产品。羧基合成法 将计量的苯加入反应釜,在4.88 MPa下通CO,于200 ℃反应4 h,一直通到CO压力不再下降,反应结束。经处理得产品。 蒽醌 基本信息 中文名称: 蒽醌 中文同义词: 蒽醌;9,10-蒽醌;9,10-蒽酮;恩菎;9,10-二氢蒽-9,10-二酮;9,10-二氧蒽;9,10-蒽醌,97%;恩 英文名称: Anthraquinone 英文同义词: MORKIT;9,10(9H,10H)-anthracenedione;9,10-Anthracenedione;9,10-anthracenequinone;9,10-Anthrachinon;9,10-dihydro-9,10-dioxo-anthracen;Anthra-9,10-quinone;Anthracene, 9,10-dihydro-9,10-dioxo- CAS号: 84-65-1 分子式: C14H8O2 分子量: 208.21 EINECS号: 201-549-0 相关类别: 化工助剂;造纸化学品;制浆过程用化学品;Intermediates of Dyes and Pigments;Chloroanthraquine, etc.;Anthraquinones;AM to AQCarbonyl Compounds;A;Alphabetic;C13 to C14;Carbonyl Compounds;Ketones;AM to AQPesticides&Metabolites;A-BAlphabetic;Alpha sort;Others;Pesticides;Pesticides&Metabolites Mol文件: 84-65-1.mol 蒽醌 性质 熔点 284-286 °C(lit.) 沸点 379-381 °C(lit.) 密度 1.438 蒸气密度 7.16 (vs air) 蒸气压 1 mm Hg ( 190 °C) 闪点 365 °F 水溶解性 <0.1 g/100 mL at 23 ºC Merck 14,687 BRN 390030 稳定性 Stable. Incompatible with strong oxidizing agents. Combustible. CAS 数据库 84-65-1(CAS DataBase Reference) NIST化学物质信息 9,10-Anthraquinone(84-65-1) EPA化学物质信息 9,10-Anthracenedione(84-65-1) 安全信息 危险品标志 Xi 危险类别码 43-36/37/38 安全说明 36/37-37/39-26-24 WGK Germany 1 RTECS号 CB4725000 毒害物质数据 84-65-1(Hazardous Substances Data) MSDS信息 提供商 语言 ACROS 英文 SigmaAldrich 英文 ALFA 中文 ALFA 英文 蒽醌 用途与合成方法 化学性质 **针状结晶。 溶于乙醇、和丙酮,不溶于水。 用途 用作染料中间体、造纸蒸煮剂及双氧水原料等 用途 用作造纸制浆蒸煮助剂,可降低用碱量,缩短蒸煮时间。 用途 蒽醌绝大部分用于染料方面,但用作制纸浆的蒸解助剂的用量已在迅速增加。(1)用于染料的生产 以蒽醌为原料,经磺化、氯化、硝化等,可得到范围很广的染料中间体,用于生产蒽醌系分散染料、酸性染料、还原染料、反应染料等,形成色谱全、性能好的染料类别。据统计,蒽醌染料有四百多个品种,在合成染料领域中占有很重要的地位。(2)用作造纸制浆蒸煮剂 在碱法蒸煮液中只需加入少量蒽醌,即可加快脱木素的速度,缩短蒸煮时间,提高纸浆得率,减少废液负荷。。蒽醌作为蒸煮添加剂的消费量增长得很快。蒽醌还有其他的应用领域。蒽醌化合物可用于高浓度过氧化氢的生产;在化肥工业中用以制造脱硫剂蒽醌二磺酸钠。在印染工业中用作拔染助剂。 生产方法 在第一次世界大战前,蒽醌产量很小,仅有以重铬酸钠将蒽氧化为蒽醌的一种生产方法。20世纪40年代发展了蒽的气相催化氧化法。在美国广泛采用苯酐法。又发展了萘醌法和苯乙烯法。1.蒽气相催化氧化法蒽氧化法是以精蒽为原料,以空气作氧化剂,五氧化二钒为催化剂,进行气相催化氧化,反应器有固定床和硫化床两种类型。 我国蒽醌生产厂大多采用固定床反应器,用含量大于90%的精蒽,熔化后用300℃左右的热空气以1560立方米/h的流速带出汽化精蒽,在热风管道中混合后通过固定床催化氧化的列管反应器,总收率达80%-85%。原料消耗定额:精蒽(90%)1260kg/t。氧化蒽醌所用原料精蒽来自煤焦油蒸馏,不含无机离子。氧化蒽醌的生产过程中,主要采用蒸馏和气相催化氧化,没有废水废气产生,不会产生氯离子、硫酸根离子和铁离子等,所以氧化蒽醌在用作高档染料方面具有竞争优势。其缺点是原料精蒽受煤焦油产品的制约。2.苯酐法 以苯酐、苯为原料,以三氯化铝为催化剂,进行付-克(Friedel-Crafts)反应,然后用浓硫酸脱水生成蒽醌。苯酐法又分为溶剂法、球磨法和气相缩合法。我国大多采用溶剂法,即以过量的苯为溶剂。此法原料易得,可以从石油做起,具有反应温度低、设备简单、副反应少等优点。缺点是污染严重,三氯化铝废酸水不易处理,而且生产成本高。我国合成蒽醌均采用苯酐法。原料消耗定额:苯酐768kg/t、纯苯700kg/t、硫酸(98%)1364kg/t、三氯化铝1554kg/t、发烟硫酸1000kg/t。3.萘醌法以萘醌和丁二烯为原料,以氯化亚铜为催化剂,进行缩合反应、脱氢后得蒽醌。由于石油化工的飞速发展,提供了此法所用的大量原料丁二烯和萘醌。该法具有消耗低、三废少等优点,在日本和美国萘醌法已达到相当规模,有发展前途。日本川崎公司使用此法生产。我国科研部门进行过大量研究,虽然小式、中式均已成功,但未工业化生产。该法的缺点是萘醌和丁二烯本身价格较高,由于反应动力学研究不够,催化剂性能不佳,经常出现床层飞温烧床,操作弹性小。4.苯乙烯法 由苯乙烯先进行二聚反应,然后氧化成邻苯酰基苯甲酸,再环合成蒽醌。该方法的优点是原料易得,没有苯酐法的铝盐废水引起的公害问题,产品成本较低。但反应条件较苛刻,技术复杂,设备要求高,是德国BASF研究的新成果。此外,日本三井化学公司获得了以甲苯为原料制备蒽醌的专利。由于工艺简单、原料便宜,引起了人们的关注。 生产方法 精蒽氧化法气相固定床氧化法 将精蒽加入气化室加热气化后与空气混合,二者比例为1︰(50~100)。混合气体进入氧化室,在V2O5催化下于(389±2) ℃下氧化,经薄壁冷凝后即得产品。液相氧化法 将精蒽计量后加入反应釜,再加入三氯苯在搅拌下溶解。然后滴加硝酸,控制反应温度105~110 ℃,将副产物NO排除,反应6~8 h后,减压蒸出溶剂,冷却结晶。得产品。此法设备腐蚀严重。苯酐法 将苯酐计量后加入反应釜,加苯在搅拌下加热熔解。加热至370~470 ℃,使混合气通过硅铝催化剂进行气相缩合。得产品。羧基合成法 将计量的苯加入反应釜,在4.88 MPa下通CO,于200 ℃反应4 h,一直通到CO压力不再下降,反应结束。经处理得产品。 类别 农药 毒性分级 低毒 急性毒性 口服- 小鼠 LD50: 5000 毫克/ 公斤; 口服-大鼠 LDL0: 15000 毫克/公斤 可燃性危险特性 明火高温可燃; 燃烧产生刺激烟雾 储运特性 库房通风低温干燥 灭火剂 干粉,泡沫,二氧化碳, 雾状水 职业标准 STEL 5 毫克/ 立方米 蒽醌 上下游产品信息 上游原料 硫酸-->苯-->三氯化铝-->苯酐-->重铬酸钠-->一氧化碳-->苯乙烯-->烟酸-->焦油-->五氧化二钒-->蒽-->三氯苯-->硅铝-->精蒽-->氧化蒽醌 下游产品 过氧化氢-->1-氨基蒽醌-->苯并蒽酮-->2-溴蒽醌-->还原绿 3-->分散兰 56-->1-氯蒽醌-->蒽酮-->1,5-二硝基蒽醌-->1,2-二羟基蒽醌-->正己酸乙酯-->直接耐晒绿 5GLL-->1-蒽醌磺酸-->2,6-二氨基蒽醌-->1,8-二硝基蒽醌-->9,10-二甲基蒽-->蒽醌-1,5-二磺酸-->1,5-二氨基蒽醌-->2-蒽醌磺酸-->2,3-二甲基蒽醌-->1,5-二氯蒽醌-->1,4,5,8-四氯蒽醌
苯环是特殊结构,介于单双键之间,不是真正双键,与烷烃类似,不能被高锰酸钾氧化,不与溴水加成。可萃取溴水中溴
苯的化学性质较稳定,对酸性KMnO4溶液和溴水均无反应。易燃,燃时有浓黑烟。苯的化学反应可分为三大类:取代反应,如硝化反应和磺化反应;加成反应,如在镍为催化剂作用下,苯跟H2反应生成环己烷;苯环破裂反应,如苯在V2O5催化剂作用和加热条件下,用空气氧化生成顺丁烯二酸酐:
通过这些反应,可由苯制成多种重要的化学中间体,它们是合成橡胶、塑料、纤维、洗涤剂、染料、医药、农药、炸药等的重要基础原料
苯
,自由的百科全书
跳转到: 导航, 搜索
苯
IUPAC中文命名
苯
常规
分子式 C6H6
SMILES C1=CC=CC=C1
分子量 78.11 g/mol
外观 无色透明易挥发液体
气味 有强烈芳香气味。12ppm浓度时可检测到油漆稀释剂气味
CAS号 71-43-2
RTECS号 CY1400000
IMDG规则页码 3185
UN编号 1114
性质
STP下的密度 0.8786 g/cm3
溶解度 0.18 g/ 100 ml 水
熔点 278.65 K (5.5 ℃)
沸点 353.25 K (80.1 ℃)
相态
三相点 278.5 ± 0.6 K
临界点 289.5℃
4.92MPa
熔解热
(ΔfusH) 9.84 kJ/mol
汽化热
(ΔvapH) 44.3 kJ/mol
燃烧热 3264.4 kJ/mol
危险性
闪点 -10.11℃(闭杯)
自燃 562.22℃
爆炸极限 1.2 - 8.0 %
摄取 可引起急性中毒,麻痹中枢神经,需要充分漱口,喝水,尽快洗胃。
吸入 可导致呼吸困难。严重者可能导致呼吸及心跳停止。
皮肤 变干燥,脱屑,皴裂,有的可能发生过敏性湿疹
眼睛 有刺激性。需用大量清水冲洗
处理方式
* 危险性:
o 遇热、明火易燃烧、爆炸。
* 人身保护:
o 防护手套,防护服,浓度过高须配带防毒面具
* 稳定性:
o 能与氧化剂强烈反应。不能与乙硼烷共存。
* 储存:
o 阴凉,通风。远离火种、热源。防止阳光直射。密封储存。防止静电
液体性质
标准生成焓
(ΔfH0液) 48.95 ± 0.54 kJ/mol
标准熵
(S0液) 173.26 J/mol·K
热容
(Cp) 135.69 J/mol·K (298.15 K)
若非注明,所有数据都依从国际单位制和来自标准温度和压力条件下。 参考和免责条款
苯(C6H6)在常温下为一种无色、有甜味的透明液体,并具有强烈的芳香气味。苯可燃,有毒,也是一种致癌物质。
化学上,苯是一种碳氢化合物也是最简单的芳烃。它难溶于水,易溶于有机溶剂,本身也可作为有机溶剂。苯是一种石油化工基本原料。苯的产量和生产的技术水平是一个国家石油化工发展水平的标志之一。苯具有的环系叫苯环,是最简单的芳环。苯分子去掉一个氢以后的结构叫苯基,用Ph表示。因此苯也可表示为PhH。
目录
[隐藏]
* 1 发现
* 2 结构
* 3 物理性质
* 4 化学性质
o 4.1 取代反应
+ 4.1.1 卤代反应
+ 4.1.2 硝化反应
+ 4.1.3 磺化反应
+ 4.1.4 烷基化反应
o 4.2 加成反应
o 4.3 氧化反应
o 4.4 其他反应
* 5 制备
o 5.1 从煤焦油中提取
o 5.2 从石油中提取
+ 5.2.1 催化重整
+ 5.2.2 蒸汽裂解
o 5.3 芳烃分离
o 5.4 甲苯脱烷基化
+ 5.4.1 甲苯催化加氢脱烷基化
+ 5.4.2 甲苯热脱烷基化
o 5.5 甲苯歧化和烷基转移
o 5.6 其他方法
* 6 分析测试方法
* 7 安全
o 7.1 毒性
o 7.2 可燃性
* 8 工业用途
* 9 苯的异构体
* 10 苯的衍生物
o 10.1 取代苯
o 10.2 多环芳烃
* 11 参看
* 12 参考文献
* 13 外部链接
[编辑]
发现
凯库勒的摆动双键
放大
凯库勒的摆动双键
苯最早是在18世纪初研究将煤气作为照明用气时合成出来的。1803年-1819年G. T. Accum采用同样方法制出了许多产品,其中一些样品用现代的分析方法检测出有少量的苯。然而,一般认为苯是在1825年由麦可·法拉第发现的。他从鱼油等类似物质的热裂解产品中分离出了较高纯度的苯,称之为“氢的重碳化物”(Bicarburet of hydrogen)。并且测定了苯的一些物理性质和它的化学组成,阐述了苯分子的碳氢比。
1833年,Milscherlich确定了苯分子中6个碳和6个氢原子的经验式(C6H6)。弗里德里希·凯库勒于1865年提出了苯环单、双键交替排列、无限共轭的结构,即现在所谓“凯库勒式”。又对这一结构作出解释说环中双键位置不是固定的,可以迅速移动,所以造成6个碳等价。他通过对苯的一氯代物、二氯代物种类的研究,发现苯是环形结构,每个碳连接一个氢。也有人提出了其他的设想:
詹姆斯·杜瓦则归纳出不同结构;以其命名的杜瓦苯现已被证实是与苯不同的另外一种物质,可由苯经光照得到。
1845年德国化学家霍夫曼从煤焦油的轻馏分中发现了苯,他的学生C. Mansfield随后进行了加工提纯。后来他又发明了结晶法精制苯。他还进行工业应用的研究,开创了苯的加工利用途径。大约从1865年起开始了苯的工业生产。最初是从煤焦油中回收。随着它的用途的扩大,产量不断上升,到1930年已经成为世界十大吨位产品之一。
[编辑]
结构
苯具有的苯环结构导致它有特殊的芳香性。苯环是最简单的芳环,由六个碳原子构成一个六元环,每个碳原子接一个基团,苯的6个基团都是氢原子。
6个p轨道形成离域大∏键的电子云
放大
6个p轨道形成离域大∏键的电子云
碳数为4n+2(n是自然数),且具有单、双键交替排列结构的环烯烃称为轮烯,苯就是[6]-轮烯。
苯分子是平面分子,12个原子处于同一平面上,6个碳和6个氢是均等的,C-H键长为1.08?,C-C键长为1.40?,此数值介于单双键长之间。分子中所有键角均为120°,说明碳原子都采取sp2杂化。这样每个碳原子还剩余一个p轨道垂直于分子平面,每个轨道上有一个电子。于是6个轨道重叠形成离域大∏键,现在认为这是苯环非常稳定的原因,也直接导致了苯环的芳香性。
[编辑]
物理性质
苯的沸点为80.1℃,熔点为5.5℃,在常温下是一种无色、有芳香气味的透明液体,易挥发。苯比水密度低,密度为0.88g/ml,但其分子质量比水重,。苯难溶于水,1升水中最多溶解1.7g苯;但苯是一种良好的有机溶剂,溶解有机分子和一些非极性的无机分子的能力很强。
苯能与水生成恒沸物,沸点为69.25℃,含苯91.2%。因此,在有水生成的反应中常加苯蒸馏,以将水带出。
在10-1500mmHg之间的饱和蒸气压可以根据安托万方程(antoine)计算:
\lg P = A - {B \over C + t}
其中:P 单位为 mmHg, t 单位为 ℃, A = 6.91210, B = 1214.645, C = 221.205
[编辑]
化学性质
苯参加的化学反应大致有3种:一种是其他基团和苯环上的氢原子之间发生的取代反应;一种是发生在C-C双键上的加成反应;一种是苯环的断裂。
[编辑]
取代反应
苯环上的氢原子在一定条件下可以被卤素、硝基、磺酸基、烃基等取代,生成相应的衍生物。由于取代基的不同以及氢原子位置的不同、数量不同,可以生成不同数量和结构的同分异构体。
苯环的电子云密度较大,所以发生在苯环上的取代反应大都是亲电取代反应。亲电取代反应是芳环有代表性的反应。苯的取代物在进行亲电取代时,第二个取代基的位置与原先取代基的种类有关。
[编辑]
卤代反应
苯的卤代反应的通式可以写成:
PhH + X_2 \to PhX + HX
反应过程中,卤素分子在苯和催化剂的共同作用下异裂,X+进攻苯环,X-与催化剂结合。
以溴为例:反应需要加入铁粉,铁在溴作用下先生成三溴化铁。
FeBr_3 + Br^- \to FeBr_4^-
PhH + Br^+ + FeBr_4^- \to PhBr + FeBr_3 + HBr
在工业上,卤代苯中以氯和溴的取代物最为重要。
[编辑]
硝化反应
苯和硝酸在浓硫酸作催化剂的条件下可生成硝基苯:
PhH + HONO_2 \to PhNO_2 + H_2O
硝化反应是一个强烈的放热反应,很容易生成一取代物,但是进一步反应速度较慢。
[编辑]
磺化反应
用浓硫酸或者发烟硫酸在较高温度下可以将苯磺化成苯磺酸。
H_2SO_4 + PhH \to PhSO_3H + H_2O
苯环上引入一个磺酸基后反应能力下降,不易进一步磺化,需要更高的温度才能引入第二、第三个磺酸基。这说明硝基、磺酸基都是钝化基团,即妨碍再次亲电取代进行的基团。
[编辑]
烷基化反应
在AlCl3催化下苯环上的氢原子可以被烷基(烯烃)取代生成烷基苯,这种反应称为烷基化反应,又称为傅-克烷基化反应。例如与乙烯烷基化生成乙苯:
PhH + C_2H_4 \to Ph\!-\!C_2H_5
在反应过程中,R基可能会发生重排:如1-氯丙烷与苯反应生成异丙苯,这是由于自由基总是趋向稳定的构型。
[编辑]
加成反应
苯环虽然很稳定,但是在一定条件下也能够发生双键的加成反应。通常经过催化加氢,镍作催化剂,苯可以生成环己烷。
C_6H_6 + 3H_2 \to C_6H_{12}
此外由苯生成六氯环己烷(六六六)的反应可以在紫外线照射的条件下,由苯和氯气加成而得。
[编辑]
氧化反应
苯和其他的烃一样,都能燃烧。当氧气充足时,产物为二氧化碳和水。
2C_6H_6 + 15O_2 \to 12CO_2 + 6H_2O
但是在一般条件下,苯不能被强氧化剂所氧化。但是在氧化钼等催化剂存在下,与空气中的氧反应,苯可以选择性的氧化成顺丁烯二酸酐。这是屈指可数的几种能破坏苯的六元碳环系的反应之一。(马来酸酐是五元杂环。)
2C_6H_6 + 9O_2 \to 2C_4H_2O_3 + 4CO_2 + 4H_2O
这是一个强烈的放热反应。
[编辑]
其他反应
苯在高温下,用铁、铜、镍做催化剂,可以发生缩合反应生成联苯。和甲醛及次氯酸在氯化锌存在下可生成氯甲基苯。和乙基钠等烷基金属化物反应可生成苯基金属化物。在四氢呋喃中氯苯或溴苯和镁反应可生成苯基格林尼亚试剂。
[编辑]
制备
苯可以由含碳量高的物质不完全燃烧获得。自然界中,火山爆发和森林火险都能生成苯。苯也存在于香烟的烟中。
直至二战,苯还是一种钢铁工业焦化过程中的副产物。这种方法只能从1吨煤中提取出1千克苯。1950年代后,随着工业上,尤其是日益发展的塑料工业对苯的需求增多,由石油生产苯的过程应运而生。现在全球大部分的苯来源于石油化工。工业上生产苯最重要的三种过程是催化重整、甲苯加氢脱烷基化和蒸汽裂化。
[编辑]
从煤焦油中提取
在煤炼焦过程中生成的轻焦油含有大量的苯。这是最初生产苯的方法。将生成的煤焦油和煤气一起通过洗涤和吸收设备,用高沸点的煤焦油作为洗涤和吸收剂回收煤气中的煤焦油,蒸馏后得到粗苯和其他高沸点馏分。粗苯经过精制可得到工业级苯。这种方法得到的苯纯度比较低,而且环境污染严重,工艺比较落后。
[编辑]
从石油中提取
在原油中含有少量的苯,从石油产品中提取苯是最广泛使用的制备方法。
[编辑]
催化重整
重整这里指使脂肪烃成环、脱氢形成芳香烃的过程。这是从第二次世界大战期间发展形成的工艺。
在500-525°C、8-50个大气压下,各种沸点在60-200°C之间的脂肪烃,经铂 - 铼催化剂,通过脱氢、环化转化为苯和其他芳香烃。从混合物中萃取出芳香烃产物后,再经蒸馏即分出苯。也可以将这些馏分用作高辛烷值汽油。
[编辑]
蒸汽裂解
蒸汽裂解是由乙烷,丙烷或丁烷等低分子烷烃以及石脑油,重柴油等石油组份生产烯烃的一种过程。其副产物之一裂解汽油富含苯,可以分馏出苯及其他各种成分。裂解汽油也可以与其他烃类混合作为汽油的添加剂。
裂解汽油中苯大约有40-60%,同时还含有二烯烃以及苯乙烯等其他不饱和组份,这些杂质在贮存过程中易进一步反应生成高分子胶质。所以要先经过加氢处理过程来除去裂解汽油中的这些杂质和硫化物,然后再进行适当的分离得到苯产品。
[编辑]
芳烃分离
从不同方法得到的含苯馏分,其组分非常复杂,用普通的分离方法很难见效,一般采用溶剂进行液-液萃取或者萃取蒸馏的方法进行芳烃分离,然后再采用一般的分离方法分离苯、甲苯、二甲苯。根据采用的溶剂和技术的不同又有多种分离方法。
* Udex法:由美国道化学公司和UOP公司在1950年联合开发,最初用二乙二醇醚作溶剂,后来改进为三乙二醇醚和四乙二醇醚作溶剂,过程采用多段升液通道(multouocomer)萃取器。苯的收率为100%。
* Suifolane法:荷兰壳牌公司开发,专利为UOP公司所有。溶剂采用环丁砜,使用转盘萃取塔进行萃取,产品需经白土处理。苯的收率为99.9%。
* Arosolvan法:由联邦德国的鲁奇公司在1962年开发。溶剂为N-甲基吡咯烷酮(NMP),为了提高收率,有时还加入10-20%的乙二醇醚。采用特殊设计的Mechnes萃取器,苯的收率为99.9%。
* IFP法:由法国石油化学研究院在1967年开发。采用不含水的二甲亚砜作溶剂,并用丁烷进行反萃取,过程采用转盘塔。苯的收率为99.9%。
* Formex法:为意大利SNAM公司和LRSR石油加工部在1971年开发。吗啉或N-甲酰吗啉作溶剂,采用转盘塔。芳烃总收率98.8%,其中苯的收率为100%。
[编辑]
甲苯脱烷基化
甲苯脱烷基制备苯,可以采用催化加氢脱烷基化,或是不用催化剂的热脱烷基。原料可以用甲苯、及其和二甲苯的混合物,或者含有苯及其他烷基芳烃和非芳烃的馏分。
[编辑]
甲苯催化加氢脱烷基化
用铬,钼或氧化铂等作催化剂,500-600°C高温和40-60个大气压的条件下,甲苯与氢气混合可以生成苯,这一过程称为加氢脱烷基化作用。如果温度更高,则可以省去催化剂。反应按照以下方程式进行:
Ph\!-CH_3 + H_2 \to Ph\!-H + CH_4
根据所用催化剂和工艺条件的不同又有多种工艺方法:
* Hydeal法:由Ashiand & refing 和UOP公司在1961年开发。原料可以是重整油、加氢裂解汽油、甲苯、碳6-碳8混合芳烃、脱烷基煤焦油等。催化剂为氧化铝-氧化铬,反应温度600-650℃,压力3.43-3.92MPa。苯的理论收率为98%,纯度可达99.98%以上,质量优于Udex法生产的苯。
* Detol法:Houdry公司开发。用氧化铝和氧化镁做催化剂,反应温度540-650℃,反应压力0.69-5.4MPa,原料主要是碳7-碳9芳烃。苯的理论收率为97%,纯度可达99.97%。
* Pyrotol法:Air products and chemicals公司和Houdry公司开发。适用于从乙烯副产裂解汽油中制苯。催化剂为氧化铝-氧化铬,反应温度600-650℃,压力0.49-5.4MPa。
* Bextol法:壳牌公司开发。
* BASF法:BASF公司开发。
* Unidak法:UOP公司开发。
[编辑]
甲苯热脱烷基化
甲苯在高温氢气流下可以不用催化剂进行脱烷基制取苯。反应为放热反应,针对遇到的不同问题,开发出了多种工艺过程。
* MHC加氢脱烷基过程:由日本三菱石油化学公司和千代田建设公司在1967年开发。原料可以用甲苯等纯烷基苯,含非芳烃30%以内的芳烃馏分。操作温度500-800℃,操作压力0.98MPa,氢/烃比为1-10。过程选择性97-99%(mol),产品纯度99.99%。
* HDA加氢脱烷基过程:由美国Hydrocarbon Research和Atlantic Richfield公司在1962年开发。原料采用甲苯,二甲苯,加氢裂解汽油,重整油。从反应器不同部位同如氢气控制反应温度,反应温度600-760℃,压力3.43-6.85MPa,氢/烃比为1-5,停留时间5-30秒。选择性95%,收率96-100%。
* Sun过程:由Sun Oil公司开发
* THD过程:Gulf Research and Development公司开发
* Monsanto过程:孟山都公司开发
[编辑]
甲苯歧化和烷基转移
随着二甲苯用量的上升,在1960年代末相继开发出了可以同时增产二甲苯的甲苯歧化和烷基转移技术,主要反应为:
甲苯歧化和烷基转移反应
这个反应为可逆反应,根据使用催化剂、工艺条件、原料的不同而有不同的工艺过程。
* LTD液相甲苯岐化过程:美国美孚化学公司在1971年开发,使用非金属沸石或分子筛催化剂,反应温度260-315℃,反应器采用液相绝热固定床,原料为甲苯,转化率99%以上
* Tatoray过程:日本东丽公司和UOP公司1969年开发,以甲苯和混合碳9芳烃为原料,催化剂为丝光沸石,反应温度350-530℃,压力2.94MPa,氢/烃比5-12,采用绝热固定床反应器,单程转化率40%以上,收率95%以上,选择性90%,产品为苯和二甲苯混合物。
* Xylene plas过程:由美国Atlantic Richfield公司和Engelhard公司开发.使用稀土Y型分子筛做催化剂,反应器为气相移动床,反应温度471-491℃,常压。
* TOLD过程:日本三菱瓦斯化学公司1968年开发,氢氟酸-氟化硼催化剂,反应温度60-120℃,低压液相。有一定腐蚀性。
[编辑]
其他方法
此外,苯还可以通过乙炔加成得到。反应方程式如下:
\rm 3CH\!\equiv\!CH \longrightarrow C_6H_6
[编辑]
分析测试方法
气相色谱和液相色谱可以检测各种产品中苯的含量。苯的纯度的测定一般使用冰点法。
对空气中微量苯的检测,可以用甲基硅油等有挥发性的有机溶剂或者低分子量的聚合物吸收,然后通过色谱进行分析;或者采用比色法分析;也可以将含有苯的空气深度冷冻,将苯冷冻下来,然后把硫酸铁和过氧化氢溶液加入得到黄褐色或黑色沉淀,再用硝酸溶解,然后通过比色法分析。或者直接用硝酸吸收空气中的苯,硝化成间二硝基苯,然后用二氯化钛溶液滴定,或者用间二甲苯配制的甲乙酮碱溶液比色定量。
[编辑]
安全
[编辑]
毒性
参看苯中毒
由于苯的挥发性大,暴露于空气中很容易扩散。人和动物吸入或皮肤接触大量苯进入体内,会引起急性和慢性苯中毒。有研究报告表明,引起苯中毒的部分原因是由于在体内苯生成了苯酚。
苯对中枢神经系统产生麻痹作用,引起急性中毒。重者会出现头痛、恶心、呕吐、神志模糊、知觉丧失、昏迷、抽搐等,严重者会因为中枢系统麻痹而亡。少量苯也能使人产生睡意、头昏、心率加快、头痛、颤抖、意识混乱、神志不清等现象。摄入含苯过多的食物会导致呕吐、胃痛、头昏、失眠、抽搐、心率加快等症状,甚至亡。吸入20000ppm的苯蒸气5-10分钟便会有致命危险。
长期接触苯会对血液造成极大伤害,引起慢性中毒。引起神经衰弱综合症。苯可以损害骨髓,使红血球、白细胞、血小板数量减少,并使染色体畸变,从而导致白血病,甚至出现再生障碍性贫血。苯可以导致大量出血,从而抑制免疫系统的功用,使疾病有机可乘。有研究报告指出,苯在体内的潜伏期可长达12-15年。
妇女吸入过量苯后,会导致月经不调达数月,卵巢会缩小。对胎儿发育和对男性生殖力的影响尚未明了。孕期动物吸入苯后,会导致幼体的重量不足、骨骼延迟发育、骨髓损害。
对皮肤、粘膜有刺激作用。国际癌症研究中心(IARC)已经确认为致癌物。
接触限值:
* 中国 MAC 40 mg/m3(皮)
* 美国ACGIH 10ppm, 32mg/m3 TWA: OSHA 1ppm, 3.2 mg/m3
毒性:
* LD50: 3306mg/kg(大鼠经口);48mg/kg(小鼠经皮)
* LC50: 10000ppm 7小时(大鼠吸入)
当然,由于每个人的健康状况和接触条件不同,对苯的敏感程度也不相同。嗅出苯的气味时,它的浓度大概是1.5ppm,这时就应该注意到中毒的危险。在检查时,通过尿和血液的检查可以很容易查出苯的中毒程度。
[编辑]
可燃性
由于苯可以在空气中燃烧,因此它一般都被定为危险化学品。例如在中华人民共和国《危险货物品名表》(GB 12268-90)中,苯属第三类危险货物易燃液体中的中闪点液体。而且由于它的挥发性,可能造成蒸气局部聚集,因此在贮存,运输时一般都要求远离火源和热源,防止静电。
由于苯的冰点比较高,在寒冷天气中运输会有困难,但是加热熔化会带来危险性。
[编辑]
工业用途
早在1920年代,苯就已是工业上一种常用的溶剂,主要用于金属脱脂。由于苯有毒,人体能直接接触溶剂的生产过程现已不用苯作溶剂。
苯有减轻爆震的作用而能作为汽油添加剂。在1950年代四乙基铅开始使用以前,所有的抗爆剂都是苯。然而现在随着含铅汽油的淡出,苯又被重新起用。由于苯对人体有不利影响,对地下水质也有污染,欧美国家限定汽油中苯的含量不得超过1%。
苯在工业上最重要的用途是做化工原料。苯可以合成一系列苯的衍生物:
* 苯与乙烯生成乙苯,后者可以用来生产制塑料的苯乙烯
* 与丙烯生成异丙苯,后者可以经异丙苯法来生产丙酮与制树脂和粘合剂的苯酚
* 制尼龙的环己烷
* 合成顺丁烯二酸酐
* 用于制作苯胺的硝基苯
* 多用于农药的各种氯苯
* 合成用于生产洗涤剂和添加剂的各种烷基苯
此外还可以用来合成氢醌,蒽醌等化工产品。
[编辑]
苯的异构体
* 杜瓦苯
* 盆苯
* 休克尔苯
* 棱柱烷
[编辑]
苯的衍生物
下面是一些有代表性的苯的取代物或与苯结构相似的物质。
[编辑]
取代苯
烃基取代
* 甲苯
* 二甲苯
* 苯乙烯
含氧基团取代
* 苯酚
* 苯甲酸
* 苯乙酮
* 苯醌
卤代
* 氯苯
* 溴苯
[编辑]
多环芳烃
* 联苯
* 三联苯
* 稠环芳烃
o 萘
o 蒽
o 菲
o 茚
o 芴
o 苊
o 薁
[编辑]
参看
* 芳香性
* BTX
* π键
* 粗苯
[编辑]
参考文献
1. 中国石化北京化工研究院,《常用危险化学品安全数据卡》(内部材料),2004年
2. 魏文德主编,《有机化工原料大全》第三卷,化学工业出版社,1994年,p358-381, ISBN 7-5025-0684-5
3. (英)汉考克(Hancock,E.G.)主编,《苯及其工业衍生物》,化学工业出版社,1982.11
4. US 3863310 (1975).
5. FR 1549188 (1972).
6. JP 45-24933 (1970).
7. GB 1241316 (1975).
8. US 3879602 (1983).
9. Wilson, L. D. "Health Hazards from aromatic Hydrocarbons", Des Plaines, III., Universal Oil Products Company, 1962
取自""
参考资料:
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。