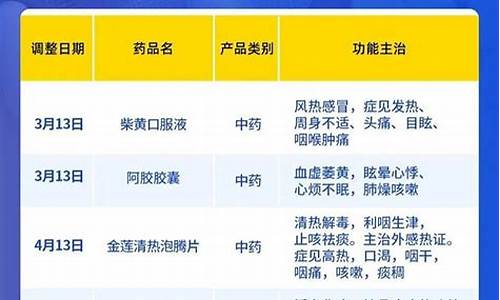
化学原料药工艺流程-化学制药原料药
我国实施纵向管理,原料药注册申报流程包括立项、研究开发、临床试验、注册申报等环节,需要经过多个部门的审批,并符合相关法律法规的要求。
我国原料药注册申报流程相对复杂,主要包括立项、研究开发、临床试验、注册申报等多个环节。立项阶段需要完成药品开发计划书、药品临床试验研究方案等申请书的编制和报送,以及对相关科研机构的审核。研究开发阶段则需要进行品种优化、指纹图谱分析、制剂工艺研究等工作,并提交质量控制资料和药物不良反应报告。临床试验阶段是个关键的环节,其实质是药品的安全性和有效性评价。药品临床试验必须按照国家临床试验管理规范进行,如《临床试验质量管理规范》、《临床试验报告内容与格式规范》等。该阶段需要提交临床试验方案、监测计划等资料,并通过相关机构的审批。临床试验完成后,还需进行数据整理、统计和分析,编制临床试验报告。注册申报是原料药上市前最后一个环节,需要向国家药品监督管理局递交注册申请,并通过审核才能获得注册证书。注册申请需要提交质量控制资料、药物不良反应报告、生产工艺及规程等内容的详细说明,并接受各级专家组的评审。
原料药注册申报时需要特别注意哪些事项?在原料药注册申报过程中,需要遵守相关药品法规,如《药品管理法》、《药品注册管理办法》等。同时,对于不同类别的原料药,还需要符合不同的管理规定,比如化学药物、生物制品等。另外,国家药品监督管理局要求申报材料必须真实准确、完整规范,如有虚假或变造情况,将面临严厉处罚。
原料药注册申报流程需要经过多个部门的审批,并符合相关法律法规的要求。申报过程中,需要编制和递交大量的申请书、方案、报告等材料,并接受各级评审。对于不同类别的原料药,还需要遵守特定的管理规定。为了顺利完成注册申报,建议咨询专业律师或顾问,确保申报过程合法合规,降低风险。
法律依据:
《中华人民共和国药品管理法》第七条 从事药品研制、生产、经营、使用活动,应当遵守法律、法规、规章、标准和规范,保证全过程信息真实、准确、完整和可追溯。
白芍中原料药白芍总苷的提取车间设记的目的是什么
1、中试——小型生产模拟试验 ?是从小试过渡到工业生产必不可少的重要环节; ?是在模型化的生产设备上(设备的设计要求和操作原 理与生产设备相同)基本完成由小试工艺向生产操作 规程(草案)的过渡; ?确保按操作规程(草案)能始终如一地生产出预定质 量标准的产品/中间体。
2、进行中试的条件 ?产品的合成路线已确定; ?小试的工艺考察已完成: ?各部反应的工艺过程及工艺参数已确定(如加料方式、 反应时间、反应温度、压力、终点控制,提取、分离、 结晶、过滤、干燥等); ?对成品的精制、结晶、分离、干燥的方法及要求已确 定(晶型、残溶); ?小试的3~5批稳定性试验说明该小试工艺可行、稳定; ?必要的材质腐蚀性试验已经完成; ?已建立原料、中间体和产品的质量控制方法/质量标准。 成熟的小试是进行中试/生产的最主要的基础
3、中试要实现的目标
目的
1、考查小试工艺的工业化生产的可能性,核对、校正和 补充实验室数据,并优化工艺条件;
2、制定生产工艺规程;为车间设计、施工安装、中间质 控以及生产管理提供必要的数据和资料。 〇物料和能量衡算,计算产品质量、经济效益、劳动强 度等
3、解决实验室阶段未能解决或尚未发现的问题;
4、为临床前的药学和药理毒理学研究以及临床试验提供 一定数量的药品。
指导原则(第二稿)对化学原料药的中试放大提出了八 项主要任务
1、考核实验室提供的工艺路线在工艺设备、条件、原材料等 方面在中试放大时是否有特殊的要求,是否适合工业化生 产;
2、确定所用起始原料、试剂或有机溶媒的规格或标准
3、验证小试工艺是否成熟合理,主要经济指标是否接近生产 要求;
4、进一步考核和完善工艺条件,对每一步反应和单元操作均 应取得基本稳定的数据;
5、根据中试研究资料制订或修订中间体和成品的质量标准、 分析方法;
6、根据原材料、动力消耗和工时等进行初步的技术经济指标 核算;
7、提出“三废”的处理方案;
8、提出整个合成路线的工艺流程,各个单元操作的工艺规程。 一般来说,中试所采用的原料、试剂的规格应与工业化生 产时一致。
求助 原料药申报
提取纯度高的白芍总苷、提高提取效率。
1、提取纯度高的白芍总苷:提取车间能够通过科学的工艺和设备,从白芍中高效地提取出纯度较高的白芍总苷。这有助于确保产品的质量和药效的稳定性。
2、提高提取效率:提取车间能够通过合理的工艺流程和设备配置,提高白芍总苷的提取效率。这有助于提高生产效率,降低生产成本。
[这个贴子最后由Jenny在 2005/07/05 09:49am 第 1 次编辑]申报资料项目:1-4、7-16,申报程序: 1:先到省安监处将该品种添加进《药品生产许可证》的生产范围。需准备的资料如下: (1)填写变更《药品生产许可证》申请表(该表格到省局网站上下载) (2)相关证明性文件: 包括:1、《药品生产许可证》正、副本复印件。 2、申请变更《药品生产许可证》的报告。 3、有关生产部门负责人简历、学历和职称证书。 4、专业技术人员、工程技术人员、技术工人登记表,高、中、初级人员比例表。 5、企业、仓储、质量检验场所平面布置图。 6、生产工艺、空气净化系统、工艺设备平面布置图。 7、有关品种质量标准及依据、品种的注册报批情况及依据。 8、有关品种的工艺流程图。 9、空气净化系统、制水系统、主要设备验证概况。 10、主要生产设备及检验仪器目录。 11、新增范围的生产、质量管理文件目录。 (3)《药品生产许可证》正、副本原件准备好以上资料,报到政务中心省局安监处(如《许可证》不变更,省局不受理该品种的注册申请,《许可证》30个工作日可变更下来,需准备好现场考核) 2:等《药品生产许可证》变更完毕后,将申报资料报注册处。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。