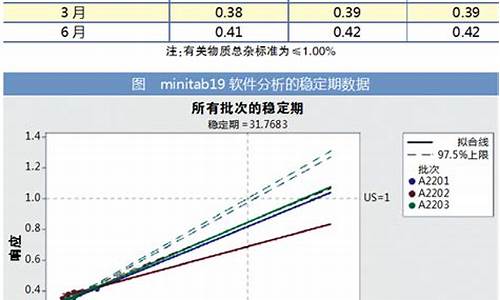
原料药的含量如规定上限为100%以上时-原料药的含量

按照ICH的相关要求和中国药典的要求,在制定药品质量标准时候应对该方法进行方法学验证。本人对其中一些原则理解不清,还请各位大虾、战友能够给予解释,谢谢。
1、关于原料药含量测定的回收率问题
(1)制剂的含量测定回收率方案容易制定,一般采用加样回收率做法。但是原料药(一般含量大于98.5%)的回收率也采用加样做法吗?使加杂质对照吗?加多少?还是用已知含量的原料配制成含量测定浓度的80%、100%和120%。后直接测定?希望能够针对滴定法、UV法和HPLC法分别距离说明下。
(2)ICH和中国药典都要求回收率是高中低三个浓度,每个浓度测定三次;请问,对于HPLC法,按三个浓度梯度,每个浓度测定三次要求,一共需要配制三份样品,每份样品进样三次,共九次,计算这九次含量结果进行评价呢还是一共配制9份样品,每个浓度梯度三份,,每份进2针,计算这9次含量结果进行评价?
2、检测限
HPLC检测限可根据信噪比确定,滴定法和UV法检测限如何做?
3、重复性试验
(1)第一种做法(3浓度/每浓度三次)
请问配制供试品液时,需要配制供试液3份,每份连续测定三次(HPLC法就是连续进样3针),这样做法是否正确,与原料药的回收率有什么不同?
(2)第二种做法(用100%浓度测定六次)
是配制1份100%浓度溶液,连续测定6次(HPLC法就是连续进样6针)还是配制6份100%溶液,每份测定一次(HPLC法就是每份进2针)?
4、准确性可在精密度、线性和专属性建立后推论而得,如何推论?
国家药品标准与企业内控质量标准为何标示量有所不同
是气相色谱法,气相色谱法用于具有一定挥发性原料药,高效液相色谱法与气相色谱法一样具有良好的的分离效果。主要用于多组分抗生素原料药和杂质干扰其他测定方法的原料药的含量测定。
原料药的含量测定,一般少用紫外光光度法,必要时,可用对照品同时测定进行比较计算,以减少不同仪器的测定误差,采用吸收系数法时,应给出E值,如此值小于100一般不宜采用。
相关要求
内标物质应选易得并不得对测定产生干扰的化学物质。所用填充剂首选十八烷基硅烷键合硅胶、硅胶、辛基硅烷键合硅胶,如经试用不合适后,再选取用其他填充剂。
流动相首选甲醇-水系统,用少量酸碱调节的流动相,或对流动相中pH敏感的品种,其流动相的pH值必须明确规定。应有“色谱条件与系统适用性试验”的要求,所订理论板数和分离度数值均指符合检测的最低要求。
含量表示意义不一样。
1、国家药品原料药一般是用百分含量表示,表示100g原料药中含多少主要成分,百分含量越接近100%,说明纯度越高。
2、企业内控制剂多用标示量百分含量表达。标示量是指每片(或粒、ml、g(软膏)等)所含主要成分的量,标示量%=真实含量/标示量*100%,理解了标示量%计算公式,就知道标示量%的意义。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。