原料药的研究方向-原料药的研究

摘要:原料药指的是用于生产各类制剂的原料药物,是无法直接服用的物质。有的人常常把它和中间体混淆,两者还是有一定不同的,原料药和中间体的区别在于定义方面、认证方面、新药开发的角度、药事管理角度等方面上。具体的原料药是指什么以及原料药和中间体的区别在哪里,一起到文中来看看吧!一、原料药是指什么
原料药,指用于生产各类制剂的原料药物,是制剂中的有效成份,病人无法直接服用的物质。
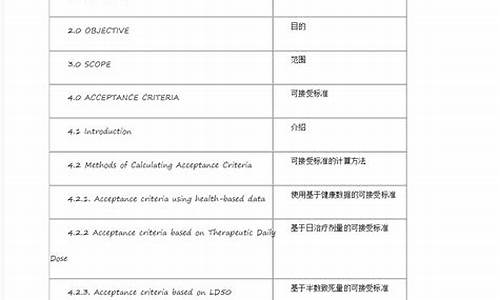
原料药在ICHQ7A中的完善定义:旨在用于药品制造中的任何一种物质或物质的混合物,而且在用于制药时,成为药品的一种活性成分。此种物质在疾病的诊断,治疗,症状缓解,处理或疾病的预防中有药理活性或其他直接作用,或者能影响机体的功能或结构。
二、原料药和中间体的区别在哪里
1、定义不同
(1)中间体:Intermediate:原料药工艺步骤中产生的、必须经过进一步分子变化或精制才能成为原料药的一种物料。中间体可以分离或不分离。
(2)原料药:ActivePharmaceuticalIngredient(API)(orDrugSubstance)_活用成分:旨在用于药品制造中的任何一种物质或物质的混合物,而且在用于制药时,成为药品的一种活性成分。此种物质在疾病的诊断,治疗,症状缓解,处理或疾病的预防中有药理活性或其它直接作用,或者能影响机体的功能和结构。
从定义中可以看出,中间体是制作原料药的前道工序的关键产物,与原料药结构不同。另外,药典中有原料药的检测方法,但是没有中间体的。
2、认证方面的区别
(1)中间体(FDA)目前FDA要求中间体必须进行注册,CEP则不用,但是CTD文件中要有中间体的详细工艺描述。而国内,对中间体没有GMP强制要求。
(2)原料药(API)由API企业提交的,如果API合成路线非常简单,如只有一步反应,FDA认为风险控制不足,就非常有可能去延伸检查中间体。中间体管理一般按照ISO或者结合Q7a,有质量体系管理就可。
3、从新药开发的角度说
(1)原料药是经过充分药学研究可以安全的用于人体起治疗诊断作用的一个化合物。
(2)中间体是合成原料药过程中的化合物,不一定具备治疗作用或者有毒性。注意,这里说的是不一定,有些原料药合成过程中的中间体也是原料药。
4、从药事管理的角度说
(1)原料药要依法向药监当局(在中国是国家食品药品监督管理总局,在美国是FDA,在欧洲是EMA)申请注册,取得批准文号后在符合GMP的厂房中合成。
(2)中间体只是合成原料药过程中的中间产物,不需要取得文号。需要说明的是与原料药同样的化合物没有取得文号或者不是在GMP厂房中生产都不是原料药。
药剂学研究的内容不包括
新摇床钱研究主要内容包括:
临床前研究:
化学部分:
原料药结构确证;
同位素内标的合成(用于药代实验);
晶型的初步研究;
杂质的初步研究;
放大工艺的稳定性研究;
安全性评价及制剂工艺研究的GLP样品生产;
分析部分:
样品的全检;
质量标准的确定;
分析方法的验证;
药理:
药效实验;
药代实验;
安全性评价实验;
制剂:
溶液稳定性研究;
处方前研究。
处方工艺研究;
处方放大稳定性研究。
原料药和制剂的光稳定性试验有什么异同?
药剂学研究的内容不包括新原料药的研究与开发。
药剂学的研究内容:
1、药剂型因素的研究。研究药物剂型因素和药效之间的关系,这里所指的剂型不仅是指片剂、注射剂、软膏剂等剂型概念。
还包括跟剂型有关的各种因素,如药物的理化性质(粒径、晶型、溶解度、溶解速度、化学稳定性等)、制剂处方(原料、辅料、附加剂的性质及用量)、制备工艺(操作条件)以及处方中药物配伍及体内相互作用等。
2、生物因素的研究。研究机体的生物因素(年龄、生物种族、性别、遗传、生理及病理条件等)与药效之间的关系。
3、体内吸收机理等的研究。研究药物在体内的吸收、分布、代谢和排泄的机理对药效的影响,保证制剂有良好的生物利用度和安全有效。
药剂学:
药剂学一般分为西药,中药制剂。它学到知识跟护理专业比较相似,比如医学的基础知识,解剖,生理这些都是要学的。
药剂学专业主要课程:有机化学、物理化学、生物化学、微生物学、药物化学、药剂学、药理学、药物分析学、药事管理学、临床医学概论。
药剂学专业学习的课程主要有:
物理化学、化工原理、药物化学、药物分析学、药理学、物理药学、药用高分子材料学、生物药剂学与药物动力学、工业药剂学、制剂设备与车间工艺设计、药品经营与管理、定量分析、仪器分析、
药事管理学、有机化学、分析化学、生物化学、药用植物学与生物学、天然药物化学、计算机基础、形态学概论、毒理学基础、药物的波普解析、医学微生物学、基础化学实验、细胞生物学、专业技能实验等课程。
为什么研究和开发分离纯化技术对于原料药生产具有举足轻重的作用
药物的稳定性:在特定的容器或密闭系统中可保持药物原有的物理、化学、生物学、治疗、毒理及其相关性能。理想的情况是药物原料的纯度、杂质含量在各个安全性实验、临床试验评估、制剂研究和稳定性实验中都保持恒定。
加速试验(Accelerated testing):加速试验是采用超出贮藏条件的试验设计来加速原料药或制剂的化学降解或物理变化的试验,是正式稳定性研究的一部分。(加速试验数据还可用于评估在非加速条件下更长时间的化学变化,以及在短期偏离标签上注明的贮藏条件(如运输过程中)时对质量产生的影响;但是,加速试验结果有时不能预测物理变化。)
中间试验或中间条件试验:中间试验是为拟在25℃下长期贮藏的原料药或制剂设计的在30℃/65%RH条件下进行的试验,目的是适当加速原料药或制剂的化学降解或物理变化。
长期试验:长期试验是为确定在标签上建议(或批准)的有效期(复检期)进行的,在拟定贮藏条件下的稳定性研究。
注册批次:用于正式稳定性研究的原料药或制剂批次,其稳定性数据在注册申报时可分别用于建立原料药和制剂的有效期(复检期)。原料药申报批次均至少是中试规模;新制剂3个批次中至少2个批次是中试规模,另1个批次的规模可小一些,但必须采用有代表性的关键生产步骤;仿制制剂申报批次均至少是中试规模。注册批次也可以是生产批次。
生产批次:使用申报时确认的生产厂房及生产设备,以生产规模生产的原料药或制剂批次。
承诺批次:注册申报时承诺的在获得批准后开始进行或继续完成稳定性研究的原料药或制剂的生产规模批次。
复检期:通常对多数已知不稳定的生物技术/生物原料药和某些抗生素,建立确认的是有效期,而对多数较稳定的化学原料药,建立确认的实为复检期。复检期是在此期间内,只要原料药保存于规定的条件下,就认为其符合质量标准,并可用于生产相应的制剂;而在此期限后,如果用该批原料药生产制剂,则必须进行质量符合性复检;如复检结果显示其质量仍符合质量标准,则应立即使用;1批原料药可以进行多次复检,且每次复检后可以使用其中的一部分,只要其质量一直符合质量标准即可。
影响因素试验(制剂):是指为评估剧烈条件对制剂质量的影响而进行的研究。该试验包括光稳定性试验和对某些制剂(如:定量吸入制剂、乳膏剂、乳剂和需冷藏的水性液体制剂)的特定试验。
提高药品质量,降低生产成本。
1、提高药品质量。研究和开发分离纯化技术,可以提高要药品的纯度,以保证药品的质量更好。
2、降低生产成本。研究和开发分离纯化技术,能够更充分的利用杂质中的有效成分,减少原料支出,降低生产成本。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。