原料药的来源有哪些方面-原料药的来源有哪些

马来酸氯苯那敏原料药被召回,这个消息对于很多人来说可能会引起担忧和困惑。作为一种常用的抗过敏药物,马来酸氯苯那敏被广泛使用于治疗过敏症状,如鼻塞、咳嗽、打喷嚏等。近期发现该药物存在一些副作用风险,因此相关企业决定对马来酸氯苯那敏原料药进行召回。
马来酸氯苯那敏原料药的副作用主要包括但不限于头晕、乏力、口干、恶心等。这些副作用可能会对患者的日常生活产生一定影响,所以我们需要保持警惕,并在使用该药物时谨慎选择。如果在使用过程中出现不适反应或副作用,请及时咨询医师。
我们要明确一点,马来酸氯苯那敏原料药的召回并不意味着该药物完全无效或危害巨大。召回是为了保障广大消费者的健康和安全,并对疑似问题产品进行检查和更换。因此,我们不必过分恐慌,但也不能掉以轻心。
作为消费者,我们在使用马来酸氯苯那敏时需要采取一些预防措施。一定要严格按照医生的建议和剂量使用该药物,避免自行增加或减少用药量。在使用过程中如有任何不适或副作用反应,应立即停止使用并咨询医师。
我们还可以尝试其他替代药物来缓解过敏症状。目前市面上有许多其他成分的抗过敏药物可供选择,如氯雷他定、扑尔敏等。在选择替代药物时,我们可以咨询医师的意见,以确保选用适合自己的药物。
我想提醒大家注意药品的购买渠道和品牌信誉。购买药品时应选择正规渠道,避免购买假冒伪劣产品。了解一下相关品牌的信誉和口碑也是很重要的参考因素。
马来酸氯苯那敏原料药的召回是出于对消费者健康和安全的考虑。作为消费者,我们要保持警惕,合理使用药物,并及时咨询医师。多了解其他替代药物也是有益的,可以帮助我们更好地选择适合自己的治疗方案。记住,健康是最重要的财富,让我们一起保护好自己的身体吧。
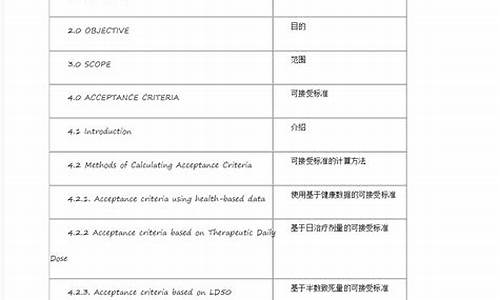
原料药和新制剂中的杂质,哪些需要对其定性或结构确证
这个说起来就有点多了。下面大概说下国产品种。
每一个药品都需要获得药监局核发的生产批文才能够生产并上市销售。这里说的生产批文包括了LZ上面说的“药品注册证、药品注册批件、药品再注册批件”等等,某个品种第一次注册的时候核发的是药品注册批件,以前还会有配套的药品注册证(现在没有这个东西了),那么只要这个“药品注册批件”在有效期内,该品种都是可以生产的。注册批件有效期只有5年,当注册批件过期了就需要再注册,一般是提前半年申请再注册,药监局受理该申请后会发一个受理通知书(这个受理通知书是没有实际意义的,仅仅表示受理该申请,至于能不能通过还需要审核),审核通过才会核发药品再注册批件,有了这个再注册批件,该品种的就又有了5年的有效期。以此类推,每隔五年都需要重新注册。药品补充申请批件是指对该品种的一些补充申请核发的批件,这里说的补充申请的内容就很多了,生产厂家更名,药品变更有效期,生产地址变更,增加规格等等,都属于补充申请的范围,需要具体问题具体分析了。综上所述,只要具备了在有效期内的生产批文,该品种就能生产。如果之前的注册批件过期了,新的再注册批件还没有下来,仅有申请再注册的受理通知书,这期间是不能生产的。
如何判断天然药物化学成分的纯度
原料药是原料药,制剂是制剂。你知道这些杂质是原料药中带入的,只能证明了它的来源。现在对于杂质的要求非常严格,即便是原料药中的杂质也分为:起始物料、中间体或者降解产物等等。
看这个吧,超过了报告限度的杂质,是需要出报告的。超过鉴定限度的杂质是需要知道它具体的化学结构的。
尤其是你在制剂研究、生产过程中监控这些杂质的增长情况。有些杂质在原料药里增长不明显,做成了制剂就蹭蹭地长。到底是湿度影响,还是酸碱度影响,或者是温度影响。这关系到制剂工艺的问题,另外产品贮藏条件,包装条件都是跟主成分含量以及杂质有关系的。,具体的可以查询药智网,回答满意请您采纳,谢谢。
原料中控制的杂质,制剂中有必要或者必须继续控制吗?依据
判断天然药物化学成分的纯度的方法是通过样品的外观如晶形以及熔点、溶程、比旋度、色泽等物理常数进行判断。
天然药物的介绍
天然药物,是指经现代医药体系证明具有一定药理活性的动物药、植物药和矿物药等。天然药物不等同于中药或中草药。
随着社会的发展,人们越来越关注化学药品给人类自身健康及生活环境带来的负面影响;回归自然、保护环境已成为一种处理人类和环境关系的潮流思想。包括植物药、动物药和海洋药物的天然药物的研究和开发顺势大力发展,对天然药物的各种人为禁制也趋于宽松。
植物药,顾名思义,是以植物初生代谢产物如蛋白质、多糖和次生代谢物如生物碱、酚类、萜类为有效成分的原料药、制剂。市场上植物来源的中药、中成药均在植物药之列。植物药在天然药物中占主导地位。
由于其在治疗上的独特优势(来自大自然,毒副作用小;在治疗疑难杂症上有广阔的前景)而倍受重视。
天然药物是指在现代医药理论指导下使用的天然药用物质及其制剂。其来源包括植物、动物和矿物,一般不包括来源于基因修饰动植物的物质、经微生物发酵或经化学等修饰的物质。
天然药物的研发应关注以下几点:一是以现代医药理论指导临床试验方案设计与评价;二是活性成份的确定应有充分的依据;三是应有充分的试验数据说明处方合理性、非临床和临床的有效性以及安全性;四是保证资源的可持续利用。
生物制药是用生物工程的方法及分离提纯工艺获得治疗疾病的有效成分。植物药为生物制药提明方向。基因工程药物提得很多;基因工程药物怎么来,把控制有效成分合成(的酶)的基因克隆,整合到受体中去令其(超)表达,得到需要的化合物。
请问,做保健食品的中药提取物,采购来源国家药监局有规定吗?为何市面上多是兽药资质的GMP车间有资格
有,需要继续控制。
原料药是原料药,制剂是制剂。你知道这些杂质是原料药中带入的,只能证明了它的来源。现在对于杂质的要求非常严格,即便是原料药中的杂质也分为:起始物料、中间体或者降解产物等等。
看这个吧,超过了报告限度的杂质,是需要出报告的。超过鉴定限度的杂质是需要知道它具体的化学结构的。
尤其是你在制剂研究、生产过程中监控这些杂质的增长情况。有些杂质在原料药里增长不明显,做成了制剂就蹭蹭地长。到底是湿度影响,还是酸碱度影响,或者是温度影响。这关系到制剂工艺的问题,另外产品贮藏条件,包装条件都是跟主成分含量以及杂质有关系的。
所有化学药品的种类
来源没有规定的。
根据2010版GMP原料药第四章规定:
第十五条 应当对每批物料至少做一项鉴别试验。如原料药生产企业有供应商审计系统时,供应商的检验报告可以用来替代其它项目的测试。
第十六条 工艺助剂、有害或有剧毒的原料、其它特殊物料或转移到本企业另一生产场地的物料可以免检,但必须取得供应商的检验报告,且检验报告显示这些物料符合规定的质量标准,还应当对其容器、标签和批号进行目检予以确认。免检应当说明理由并有正式记录。
第十七条 应当对首次采购的最初三批物料全检合格后,方可对后续批次进行部分项目的检验,但应当定期进行全检,并与供应商的检验报告比较。应当定期评估供应商检验报告的可靠性、准确性。
1、未在国内外上市销售的药品:
(1)通过合成或者半合成的方法制得的原料药及其制剂;
(2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂;
(3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂;
(4)由已上市销售的多组份药物制备为较少组份的药物;(5)新的复方制剂。
2、改变给药途径且尚未在国内外上市销售的制剂 。
3、已在国外上市销售但尚未在国内上市销售的药品:
(1)已在国外上市销售的原料药及其制剂;
(2)已在国外上市销售的复方制剂 ;
(3) 改变给药途径并已在国外上市销售的制剂 。
4、改变已上市销售盐类药物的酸根、碱基(或者金属元素),但 不改变其药理作用的原料药及其制剂 。
5、改变国内已上市销售药品的剂型,但不改变给药途径的制 剂 。
6、已有国家药品标准的原料药或者制剂。
扩展资料
结构明确的具有预防、治疗、诊断疾病,或为了调节人体功能、提高生活质量、保持身体健康的特殊化学品。化学药物是以化合物作为其物质基础,以药效发挥的功效(生物效应)作为其应用基础的。
结构确证:
1、说明结构确证测试样品的来源(精制)和纯度
2、对照品的来源:是否合法
3、对含多个手性中心的原料药需确定本品的立体结构,长要求补充进行NOE谱 或其他图谱的测定,或提供本品详细的同样测试条件下的核磁共振文献图谱和数据以进一步确定本品的立体结构。
4、晶型研究发补原则。对于难溶性化合物,制剂为口服固体制剂,同时从文献报道已知晶型对生物利用度或稳定性有明显影响,这种情况应在临床研究前要求进行晶型研究,其它情况可不要求。
百度百科-化学药品
百度百科-化学药物
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。