原料药质量控制题目-原料药质量标准制定

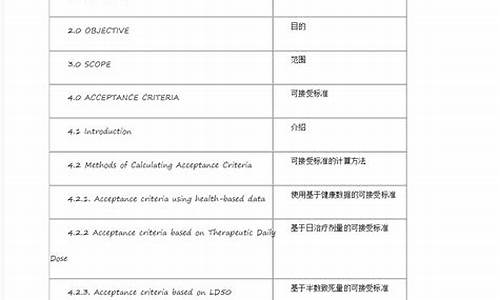
采用高效液相色谱法测定化学原料药时,论证方法的专属性为:需要验证的检测项目检测项目是为控制药品质量,保证安全有效而设定的测试项目。
根据检测项目的设定目的和验证内容的不同要求,本指导原则将需验证的检测项目分为鉴别、杂质检查(限度试验、定量试验)、定量测定(含量测定、溶出度、释放度等)、其他特定检测项目等四类。
测定方式分析
鉴别判定被分析物是目标化学原料,而非其它物质,用于鉴别的分析方法要求具有较强的专属性。杂质检查主要用于控制主成分以外的杂质,如有机杂质、无机杂质等。
而其中杂质检查可分为限度试验和定量试验两种情况。用于限度试验的分析方法验证侧重专属性和检测限。用于定量试验的分析方法验证强调专属性、准确度和定量限。
至少检查一个最小包装的是
题主是否想询问“原料药生产车间非洁净区可以使用吗”?不可以。原料药生产车间需要按照药品GMP认证标准进行建设和管理,分为不同级别的洁净区,而洁净区主要是为了控制空气微生物和粉尘等杂质的污染,在药物生产和质量控制中起着非常重要的作用,而非清洁区在生产原料时会进入粉尘等杂质的污染,所以是不可以使用的。
医药企业的药品(包括原料药和成品药)DMF怎么写?格式是怎样的?请问谁能告诉我啊?先谢谢大家了!
答案:C
同一批号的药品应当至少检查一个最小包装,但生产企业有特殊质量控制要求或者打开最小包装可能影响药品质量的,可不打开最小包装;破损、污染、渗液、封条损坏等包装异常及零货、拼箱的,应当开箱检查至最小包装;外包装及封签完整的原料药、实施批签发的生物制品,可不开箱检查。故此题选C。
原料药与药物制剂的质量控制在方法的选择上有什么异同?
DMF是英文DRUG MASTER FILE的简称,译为"药品主文件,"它是反映药品生产和质量管理方面的一套完整的文件资料。主要包括生产厂简介、具体质量规格和检验方法、生产工艺和设备描述、质量控制和质量管理等方面的内容。
根据不同国家和地区对注册程序的规定和DMF的编写要求不同,DMF大致分为两种,一种是欧洲共同体国家所要求的DMF(简写为EDMF),一种是美国FDA所要求的。前一种要求重点介绍产品的工艺质量控制、杂质和稳定性研究等方面的资料和数据;后一种DMF被细分为五类,在EDMF基础上,尚需介绍生产厂的厂房、设施、人员、GMP管理、机构和职责等方面的内容。
在欧共体,DMF是办理市场销售许可证的一部分。药品要在欧共体或销售国家药品管理局申报一套资料,办理市场销售许可证。当药品所用的活性成份(即原料药)的供应商改变时,同上办理。而DMF是申报资料的重要部分。不按要求提供DMF,就不能把所生产的产品销售到该国家。
在美国,虽然FDA没在正式文件中规定出口到美国的原料厂家必须上报DMF资料,但实际上大家都在做,而且美国FDA也发表了编写DMF文件的指南。若该原料药被用做处方药的成分时,则美国FDA一定派员对生产厂家进行检查,以确定该厂的生产是否与上报资料所述相符,是否是按美国CGMP(现行GMP)要求进行。鉴于欧共体和美国对进口原料药的严格的管理,编写一份符合要求的DMF文件对促进原料药的出口是至关重要的。
我公司自20世纪90年代初组织人员对主要出口原料药编写DMF,当时主要是按美国格式编写的。这些文件对当时我公司国际贸易的开展起了重要的作用,也使大家了解了DMF文件对原料药出口的重要性。随着国际贸易的深入和GMP的不断发展,对DMF的内容不断提出了新的要求。自1996年以后,陆续对老版本的DMF进行了改版。我公司大部分的原料药销往欧美两个市场,因此要准备两个版本的DMF文件。EDMF有固定的格式,但在内容的深度和广度上不同的客户会提出不同的要求,因此,一个产品可能会有一个以上的EDMF版本。美国DMF没有固定的格式,不同的咨询官会有不同的风格,而且,咨询官为保证能一次通过FDA的审查,都比较坚持自己固有的风格。若在编写DMF的过程中更换了咨询官,所有的资料可能会从头再来。例如:氢化可的松的美国版DMF,已经由第一个咨询官逐页审核,准备呈递DMF时,由于更换咨询官,我公司不得不将厚达2寸的DMF文件按第二位咨询官的要求改版。因此,DMF的编写不会因编好了一个版本就一劳永逸了,需要按客户的要求以及工艺和设施变更的情况不断地修改补充完善,重大的变更必须通知客户。美国FDA要求,即使没有变更,每年也需要递交一份没有改变的声明。DMF在修改较多时必须要换版。
在药典上可以清晰的看出,由于制剂可能有辅料的干扰,所以一般尽量用高效液相来进行含量测定,而原料则可以用滴定法或紫外、荧光等来检测含量。但对于那些杂质紫外吸收和药品紫外吸收波长相同的原料也要用高效液相色谱法。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。