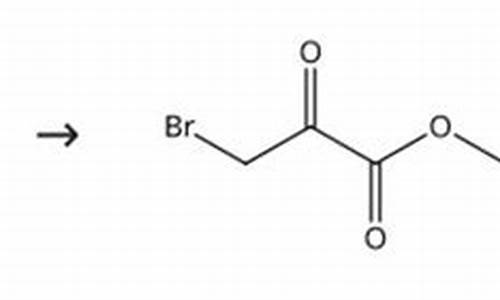
氯溴异氰尿酸和噻霉酮混用吗-溴丙酮酸乙酯和硫脲反应合成噻唑环吗
1. _ 掌握羧基的结构和羧酸的化学性质
2._掌握诱导效应和共轭效应对羧酸酸性的影响
3._ 掌握羧酸的制备方法
4, 了解重要的羧酸的主要用途
5._ 了解二元羧酸取代羧酸的特性反应
学习要求
学习内容
羧酸化合物的简介
羧酸的分类,命名和结构
羧酸的物理性质和光谱性质
羧酸的化学性质
羧酸的来源和制备
重要的一元羧酸
二元羧酸
取代酸
酸碱理论
化学性质一览表
羧酸可看成是烃分子中的氢原子被羧基(-COOH)取代而生成的化合物.其通式为RCOOH.羧酸的官能团是羧基.
布洛芬
阿司匹林
羧酸是许多有机物氧化的最后产物,它在自然界普遍存在(以酯的形式),在工业,农业,医药和人们的日常生活中有着广泛的应用.
羧酸化合物的简介
故羧基的结构为一 P-π共轭体系
_
第一节 羧酸的分类,命名和结构
一,结构
当羧基电离成负离子后,氧原子上带一个负电荷,更有利于共轭,故羧酸易离解成负离子
_
由于共轭作用,使得羧基不是羰基和羟基的简单加合,所以羧基中既不存在典型的羰基,也不存在着典型的羟基,而是两者互相影响的统一体.
羧酸的性质可从结构上预测,有以下几类:
还原反应
二,命名
1,俗名
酒石酸 马来酸
蚁酸,
蚁酸 安息香酸 草酸 琥珀酸(丁二酸)
柠檬酸(3—羟基—3—羧基戊二酸)
肉桂酸(3—苯基丙烯酸)
()
a.含羧基的最长碳链作为母体,按照主链碳原子数目命名为'某'酸 .
b.编号.从羧基C原子开始编号.(用阿拉伯数字或希腊字母.)
c. 如有不饱和键要标明烯(或炔)键的位次.并主链包括双键和叁键.将取代基的位次,数目,名称依次写在母体名称前面.,数目,名称依次写在母体前面
d. 脂环族羧酸.简单的在脂环烃后加羧酸二字,复杂的环可作为取代基.
e.芳香酸可作脂肪酸的芳基取代物命名.
f.多元羧酸:选择含两个羧基的碳链为主链,按C原子数目称为某二酸.
2,系统命名法
-乙氧基乙酸
4-甲基-4-苯基-2-戊烯酸
丙醛酸
(3-氧代丙酸或3-羰基丙酸)
3-丁酮酸
(3-氧代丁酸或乙酰乙酸)
(1R, 3R)-1,3-环己烷二羧酸
三,分类
1.按烃基的种类可分为:
a.脂肪族羧酸:饱和羧酸,不饱和羧酸
b,脂环族羧酸
c,芳香酸
2.按羧基数目可分为:一元羧酸,二元羧酸,多元羧酸_
_
饱和酸
不饱和酸
芳香酸
一元酸
乙酸
丙烯酸
苯甲酸
二元酸
乙二酸
顺丁烯二酸
邻二苯甲酸
溶解度
羧酸的物理性质
物态:C1~C3 有刺激性酸味的液体,溶于水.
C4~C9 有酸腐臭味的油状液体,难溶于水.
> C9 腊状固体,无气味.
二元羧酸,芳酸为晶体 .
羧酸是极性分子,能与水形成氢键,故低级一元酸(C1~C4)可与水互溶,但随分子量↑,在水中的溶解度↓,从正戊酸开始在水中的溶解度只有3.7 %,>C10的羧酸不溶于水.二元酸易溶于水,芳酸的溶解度也很小.苯甲酸的溶解度为 0.34g / 100gH2O
熔,沸点
①熔点:一元羧酸从C6开始,随分子量↑,呈锯齿形上升.偶数碳原子羧酸的m.p>相邻两个同系物的m.p.出现熔点双曲线.主要是偶数碳的对称性高,分子在晶体中排列整齐,晶格能较大,熔点较高.
②沸点
直链饱和一元羧酸的沸点较分子量相近的醇要高.如:甲酸,乙醇分子量均为46,沸点为100.5℃,78.3℃;乙酸,丙醇分子量为60,沸点为117.9℃,97.2℃.
主要原因为:羧酸以氢键彼此缔合, a) 此键键能大于醇之间氢键的键能.(酸中的氢键键能: 30kJ / mol ,醇中氢键键能:25kJ / mol .)b) 低级酸在蒸汽中也是以二聚体存在,所以沸点高.
IR:反映出-C=O和-OH的两个官能团
RCH2COOH R2CHCOOH
1HNMR:RCOOH
羧酸的光谱性质
羧酸的化学性质
由于共轭作用,使得羧基不是羰基和羟基的简单加合,所以羧基中既不存在典型的羰基,也不存在着典型的羟基,而是两者互相影响的统一体.
羧酸的性质可从结构上预测,有以下几类:
一,酸性
二,羧基上的羟基(OH)的取代反应
三,脱羧反应
四,α-H的卤代反应
五,羧酸的还原
羧酸的酸性比水,醇强,甚至比碳酸的酸性还要强.
羧酸离解后生成的RCOO- 负离子,由于共轭效应的存在,氧原子上的负电荷则均匀地分散在两个原子上,因而稳定,容易生成.
一,酸性
羧酸的酸性表现在:
羧酸能与碱作用成盐,也可分解碳酸盐.
此性质可用于醇,酚,酸的鉴别和分离,不溶于水的羧酸既溶于NaOH也溶于NaHCO3,不溶于水的酚能溶于NaOH不溶于NaHCO3,不溶于水的醇既不溶于NaOH也溶于NaHCO3.含羧基的有机物,在碱中可增加水溶性.如:青霉素G是含羧基的有机物,不溶于水.一般制成钾钠盐增加水溶性,易于吸收.
影响羧酸酸性强度的因素
1,电子效应对酸性的影响
2,取代基位置对苯甲酸酸性的影响
3,场效应的影响
1,电子效应对酸性的影响
1°吸电子诱导效应使酸性增强.
FCH2COOH > ClCH2COOH > BrCH2COOH > ICH2COOH > CH3COOH
pKa值 2.66 2.86 2.89 3.16 4.76
2°供电子诱导效应使酸性减弱.
CH3COOH > CH3CH2COOH > (CH3)3CCOOH
pKa值 4.76 4.87 5.05
3°吸电子基增多酸性增强.
ClCH2COOH > Cl2CHCOOH > Cl3CCOOH
pKa值 2.86 1.29 0.65
1)诱导效应
2) 共轭效应 当羧基能与其他基团共轭时,则酸性增强
4°吸电子基的位置距羧基越远,酸性越小.
CH3COOH +C
④ (OH) -I RCH2OH > R2CHOH > R3COH
醇相同时 HCOOH > CH3COOH > RCH2COOH > R2CHCOOH > R3CCOOH
(3) 成酯方式
酯化时,羧酸和醇之间脱水可有两种不同的方式:
究竟按哪种方式脱水,与羧酸和醇的结构及反应条件有关.经同位素标记醇的办法证实:
Ⅰ 伯醇和仲醇与羧酸的酯化是按酰氧键断裂进行的.
Ⅱ 叔醇与羧酸的酯化是按烷氧键断裂进行的.
H2O中无O18,说明反应为酰氧断裂.
(4)酯化反应历程
1°,2°醇为酰氧断裂历程,
3°醇(叔醇)为烷氧断裂历程.
CH3COOH + SOCl2 CH3COCl + SO2 + HCl
亚磷酸不易挥发,故该法适用于制备低沸点酰氯.
磷酰氯沸点较低(105.3℃),故适用于制备高沸点酰氯
该法的副产物均为气体,有利于分离,且产率较高.
2.酰卤的生成
羧酸与PX3,PX5,SOCl2作用则生成酰卤.
因乙酐能较迅速的与水反应,且价格便宜,生成的乙酸有易除去,因此,常用乙酐作为制备酸酐的脱水剂.
1,4和1,5二元酸不需要任何脱水剂,加热就能脱手生成环状(五元或六元)酸酐.
3.酸酐的生成
羧酸在脱水剂作用下加热,脱水生成酸酐.
不对称酸酐用羧酸盐与酰氯反应制备
例如:
_
二元酸的二铵盐受热则发生分子内脱水兼脱氨,生成五元或六元环状酰亚胺.
4.酰胺的生成
在羧酸中通入氨气或加入RNH2,R2NH ,可得到羧酸铵盐,铵盐热解失水而生成酰胺.
_
_
三,脱羧反应
羧酸在一定条件下受热可发生脱羧反应.
饱和一元羧酸在加热下较难脱羧,但低级羧酸的金属盐在碱存在下加热则可发生脱羧反应.
洪塞迪克尔(Hunsdiecker)反应:羧酸的银盐在溴或氯存在下脱羧生成卤代烷的反应.
_此反应可用来合成比羧酸少一个碳的卤代烃.
羧酸与 HgO + Br2 也可得卤烃,称为 克利斯脱反应
羧酸与 (CH3COO)4Pb ·LiCl得氯代烃 称为 柯奇反应
一元羧酸的α碳原子上连有-NO2,-C≡N,
-CO-,-Cl 等强吸电子集团时,易发生脱羧.
某些芳香族羧酸不但可以脱羧,且比饱和一元酸容易.
现可采用气相催化脱羧有羧酸直接来制备酮.
电解羧酸盐溶液可在阳极发生烷基的偶合,生成烃,
该反应称为Kolbe反应.
Kolbe反应用于二元酸单酯电解生成长链二元酸酯也
是成功的例子之一.
脂肪族羧酸的α- 氢原子也可被卤原子取代,但其反应活性要比醛,酮低的多,通常要在少量红磷,硫等催化剂存在下方可进行.
控制条件,反应可停留在一取代阶段.
四,α-H的卤代反应
α-卤代酸很活泼,可以进行亲核取代反应和消除反应.如:
羧酸不易被还原.但在强还原剂LiAlH4作用下,羧基可被还原成羟基,生成相应的1°ROH
该法不仅产率高,而且不影响C=C和C≡C的存在,可用于不饱和酸的还原.
五,羧酸的还原
乙硼烷也可将羧基还原为伯醇
羧酸的来源和制备
来源: 羧酸广泛存在与自然界,常见的羧酸几乎都有俗名.自然界的羧酸大都以酯的形式存在于油,脂,(高级脂肪酸甘油酯)蜡(高级脂肪酸高级一元醇酯)中.油,脂,蜡水解后可以得到多种羧酸的混合物.
制法:
一,氧化法
二,羧化法
三,水解法
_
(一)烃的氧化——有α-H的芳烃才能氧化为苯甲酸
(二) 伯醇或醛的氧化——制备同碳数的羧酸
一.氧化法
甲基酮氧化——制备减少一个碳原子 的羧酸
(四) 烯烃,炔烃的氧化——适用于对称烯烃,炔烃和末端烯烃,炔烃
(五) 无α— H 的醛在浓碱中加热,可得酸和醇
环酮可被氧化为内酯,进而被氧化为二酸
(三).酮的氧化
二,羧化法
(一) Grignard试剂与CO2作用——制备增加一个碳原子的羧酸
(二)烯烃羰基化法——制备增加一个碳原子的羧酸
烯烃在Ni(CO)4催化剂的存在下吸收CO和H2O而生成羧酸.
1°,2°,3°RX都可使用.但乙烯式卤代烃难反应.
三,水解法
此法仅适用于1°RX(2°,3°RX 与NaCN作用易发生消除反应).
_
1._
(二)羧酸衍生物的水解
油脂和羧酸衍生物得羧酸,及副产物甘油和醇.
(三)通过乙酰乙酸乙酯,丙二酸二乙酯合成各种羧酸.
(四) Kolbe-Schmitt反应——制备增加一个碳原子酚酸
(一) 腈的水解——制备增加一个碳原子的羧酸
重要的一元羧酸
甲酸
1.结构
2.特性
① 甲酸的酸性显著高于其它饱和一元酸
② 甲酸具有还原性,能发生银镜反应.
③ 甲酸也能使高锰酸钾溶液退色.
④ 甲酸具有杀菌力,可作消毒或防腐剂.
⑤ 甲酸与浓硫酸加热,则分解生成一氧化碳和水.
乙酸 ,苯甲酸
3. 制法
甲酸的水溶液不能用蒸馏的方法得到纯甲酸,要用
无水甲酸钠加入含硫酸的甲酸中蒸馏得到.或
一,物理性质
1.物态 二元羧酸都是固态晶体,熔点比相近分子量的一元羧酸高得多.随碳原子数目的增加,熔点呈下降趋势,偶数碳比奇数为高.
2.溶解度 比相应的一元酸大,易溶于乙醇,难溶于其他有机溶剂.
二,二元羧酸的化学性质
三,重要的二元羧酸
乙二酸(草酸)具有还原性,易被氧化成二氧化碳和水.
己二酸
丁烯二酸
苯二甲酸
二元羧酸
二,二元羧酸的化学性质
1.具有羧酸的通性
对酸性而言 Ka1 > Ka2或 pKa1〈 pKa2
对于顺与反式丁烯二酸
对于酸性:Ka1(顺) 〉Ka1(反) ; Ka2(反)〉Ka2(顺)
顺反式结构在其他物理性质方面也有差异,如:水溶性顺式大于反式(顺式偶极矩大),熔点反式高于顺式(反式对称性高,晶格能大)
2.二元羧酸受热反应的规律
Blanc规则(布朗克) :在可能形成环状化合物的条件下,总是比较容易形成五元或六元环状化合物(即五,六元环容易形成).
(1) 乙二酸,丙二酸受热脱羧生成一元酸
(2)丁二酸,戊二酸受热脱水(不脱羧)生成环状酸酐
(3)己二酸,庚二酸受热既脱水又脱羧生成环酮
3.与二元醇反应
二元酸与二元醇反应可生成环酯(但仅限于五元环或六元环)
也可以生成聚酯.
(1) 乙二酸,丙二酸受热脱羧生成一元酸,
_
(2)丁二酸,戊二酸受热脱水(不脱羧)生成环状酸酐,
(3)己二酸,庚二酸受热既脱水又脱羧生成环酮,
取代羧酸
羧酸分子中烃基上的氢原子被其他原子或子团取代后形成的化合物称为取代酸.
取代酸有卤代酸,羟基酸,氨基酸,羰基酸等,其中卤代酸,氨基酸将在有关章节中讨论,这里只讨论羟基酸和羰基酸.
一,羟基酸
1.制法
2.羟基酸的性质
3.重要的羟基酸 (自学)
二, 羰基酸
分子中含有羰基,有含有羧基的化合物称为羰基酸,如丙酮酸,3-丁酮酸等.
1)卤代酸水解 用碱或氢氧化银处理α,β,γ等卤代酸时可生成对应的羟基酸.
3)列佛尔曼斯基(Reformatsky)反应 制备β-羟基酸的方法.
2) 氰醇水解 制α-羟基酸
1,羟基酸制法
_
具有醇和酸的共性,也有因羟基和羧基的相对位置的互相影响的特性反应.主要表现在受热反应规律上.
_
β-羟基酸受热发生分子内脱水,主要生成α-β
不饱和羧酸.
_
α-羟基酸受热时,两分子间相互酯化,生成交酯.
2.羟基酸的性质
γ-和δ-羟基酸受热,生成五元和六元环内酯.
α-和β-羟基酸还有被氧化后再脱羧的性质
α-和β-羟基酸的降解反应:
这是制备高级脂肪醛酮的方法
_____ 自然界中的羟基酸
① 乳酸:
结构:
存在:酸牛奶(外消旋),蔗糖发酵(左旋的),肌肉中(右旋的).
用途:具有很强的吸湿性;工业上作除钙剂(钙盐不溶于水);食品工业中作增酸剂;钙盐可补钙.
② 苹果酸(α-羟基酸)
结构: _
存在:未成熟的果实内;植物的叶子中;自然界中存在的是左旋体.
用途:制药和食品工业.
③ 洒石酸
结构
_
存在:多种水果中;或以盐的形式存在于水果中.
用途:可用作酸味剂,其锑钾盐有抗血吸虫作用.
④ 柠檬酸
结构:
_
存在:多种植物的果实中;动物组织与体液中,为无色晶体.
用途: 食品工业的调味品(有酸味),也用于制药业.
_
注意:羟基与羧基间的距离大于四个碳原子时,受热则生成长链的高分子聚酯.
α-和β-羟基酸还有羟基被氧化后再脱羧的性质.
讨论: 写出下列反应的产物℃
讨论:下列反应的产物是
1,羰基酸具有羰基和羧酸的典型反应.
2,_ 酮酸的特性反应
α-酮酸与稀硫酸共热时,脱羧生成醛.
β-酮酸受热易脱羧生成酮._
_
二, 羰基酸
酸碱理论
一,布伦斯特酸碱理论
凡是能释放质子的任何分子或离子都是酸.
布伦斯特认为酸碱强度可根据电离常数来比较
HAc Ka=1.7*10-5
H2O Ka=1.8*10-16
二,路易斯酸碱理论
路易斯酸是电子对的接受体
路易斯碱是电子对的给予体
化学性质一览表
酸性与成盐
2. 酸酐的生成
1. 酰卤的生成
羧酸衍生物的生成
如:
α-H 卤代反应
脱 羧 反 应
4. 酰胺的生成
3. 酯的生成
对羟基苯甲酸的用途
卤化反应是指用卤素原子去取代其他物质中的活性原子或原子团,得到含卤素原子的化合物。
在合成中,卤化反应通常起桥梁的作用,R-X(代表含有卤素原子的有机物)可以的得到相同碳原子数的醇,进而得到醛酮羧酸,也可以得到腈,从而得到多一个碳原子的羧酸或胺,此外,苯环上 的烷基还原反应也和卤素原子有很大关系。
希望对你有所帮助!
不懂请追问!
望采纳!
组合化学的定义及特点
对羟基苯甲酸是用途广泛的有机合成原料,特别是其酯类,包括对羟基苯甲酸甲酯(尼泊金甲)、乙酯(尼泊金乙)、丙酯、丁酯、异丙酯、异丁酯,可做食品添加剂,用于酱油、醋、清凉饮料(汽水除外)、果品调味剂、水果及蔬菜、腌制品等,还广泛用于食品、化妆品、医药的防腐、防霉剂和杀菌剂等方面。对羟基苯甲酸也用作染料、农药的中间体。在农药中用于合成有机磷杀虫剂GYAP、CYP;在染料工业中用于合成热敏染料的显色剂;还可用于彩色胶片及合成油溶性成色剂“538”及尼龙12中用作增塑剂的生产原料。另外,还用于液晶聚合物和塑料。
作防腐剂、杀菌剂。药理实验表明,对小鼠的眼镜蛇中毒有明显的保护作用。本品可抑制霉菌的生长,与乙醇、丙醇、丁醇等醇类反应生成的各种酯类,是优良的防腐剂。本品还可用于染色、有机合成工业等领域作防腐剂、杀虫剂。
一、 在防腐剂方面的应用
1、尼泊金酯类
目前,对羟基苯甲酸制备的酯类是其消耗最大的用途,又称尼泊金酯。尼泊金酯类种类较多,从尼泊金甲酯到庚酯,在理论上还可有更长碳链的酯类。上世纪20年代,首次报道了尼泊金酯类的抗菌活性,1923年尼泊金酯类就被建议为食品和药品的防腐剂,1923年尼泊金酯正式被批准应用于食品中。后来又应用于化妆品、医药等领域,是目前应用最广泛的防腐剂之一。
我国规定食品添加剂使用卫生标准(GB2760-2007)规定,对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯及其钠盐可以应用于多种食品中,用量在用量在0.012-0.5g/kg之间(以对羟基苯甲酸计)。我国台湾省《食品添加剂使用范围及用量标准》规定:对羟基苯甲酸乙酯、丙酯、丁酯、异丙酯、异丁酯,用量在0.012-0.4g/kg之间(以对羟基苯甲酸计)。
美国、欧洲各国主要使用尼泊金甲酯、乙酯和丙酯,庚酯在美国也能应用于饮料酒中;日本使用的主要是丁酯。
大多数国家在使用对羟基苯甲酸酯类作为食品防腐剂时,一般都是将不同的酯类混合使用 ,取其协同作用,以提高防腐效果。由于对羟基苯甲酸酯类溶于氢氧化钠溶液而形成对羟基苯甲酸酯钠,水溶性增高,但储藏稳定性降低。
尼泊金酯类是各国都认可的、传统的、无刺激、不致敏和安全的化妆品防腐剂,其用量在化妆品防腐剂中一直占据前列。在我国2007年版的《化妆品卫生规范》中,可以使用的尼泊金酯类防腐剂的上限为单一酯:0.4%(以酸计),混合酯:0.8%(以酸计)。规范中,没有列举具体尼泊金酯类。根据其检测方法中描述至少有尼泊金甲酯、乙酯、丙酯、异丙酯、丁酯、异丁酯等。
在药品中作为防腐辅料使用的尼泊金酯类主要有尼泊金乙酯(英国、中国、欧洲、法国、德国、日本、瑞士、美国、国际药典),尼泊金丙酯(奥地利、比利时、英国、捷克、法国、德国、意大利、日本、荷兰、葡萄牙、瑞士、美国、欧洲、国际药典),尼泊金丙酯钠(意大利、西班牙、英国、意大利、美国、澳大利亚、法国、德国药典),尼泊金丁酯(英国、法国、德国、日本、瑞士、美国、欧洲药典),尼泊金丁酯钠(英国药典),尼泊金苄酯(英国、国际药典)。
尼泊金酯与目前的几种化学防腐剂相比,有;用量较少、成本较低、安全性好、抑菌范围广、在较宽PH值(4-8)内有效等优点,还可以多种酯混合或和相应尼泊金酯钠盐复配,不仅可以提高溶解度,还由于它们之间存在协同作用,所以具有更好的防腐能力。
尼泊金酯类的合成和一般的酯相似,由对羟基苯甲酸和相应的醇在酸性催化剂的作用下发生酯化反应。最传统、普遍的方法就是对羟基苯甲酸和相应的醇在硫酸的催化下,利用甲苯作为带水剂,酯化得到相应的尼泊金酯,收率可以达到95%以上。但是利用硫酸做催化剂的缺点也很明显:反应时间长,醇、硫酸用量大;硫酸的强氧化性易使产品颜色变深,还会引起醚化、氧化、磺化等副反应;硫酸的强酸性还严重腐蚀仪器设备;催化剂硫酸不能回收使用,排出的废酸、废水污染环境。近些年来,针对硫酸催化剂的缺点,开发了多种催化剂,使尼泊金酯的收率和品质都大有提高[2]。
2、其他防腐杀菌物质
除了尼泊金酯类业已成熟,并实际应用的防腐剂,科研工作者还应用对羟基苯甲酸合成了其他类似的可以防腐杀菌的物质。赵希荣和夏文水[3]在适宜的反应条件下成得到了对羟基苯甲酸壳聚糖酯,该酯溶解性略优于对羟基苯甲酸庚酯,而醇溶性显著提高;对大肠杆菌和金**葡萄球菌抗菌试验表明,该酯抗菌活性大于对羟基苯甲酸庚酯,更优于壳聚糖。
二、 在香精工业中的应用
对羟基苯甲酸和硫酸二甲酯反应可制得对甲氧基苯甲酸,又名对茴香酸、大茴香酸,主要用于制作香料,也有一定的防腐杀菌的性能。对甲氧基苯甲酸和乙醇在酸的催化下酯化得到的茴香酸乙酯,呈淡的水果和茴香香气,是天然等同香料和人造香料,用于配制茴芹、小茴香、甘草等型香精。
对羟基苯甲酸也可制得对乙氧基苯甲酸,具有和对甲氧基苯甲酸相似的性能。
三、在药物合成中的应用
对羟基苯甲酸除了合成尼泊金酯作为防腐剂用于医药制剂中外,还可以作为多种医药产品的基础原料。
1、非布索坦[4]
非布索坦是新一代黄嘌呤氧化酶抑制剂,临床上用于治疗尿酸过高症(痛风),帝人公司于04年年初在首先日本上市。
非布索坦的合成:以对羟基苯甲酸甲酯为原料,经过溴化、醚化得到关键中间体3-溴-4-(2-甲基丙氧基)苯甲酸甲酯,再与氰化亚铜反应引入氰基,然后合成噻唑环,最后经水解得到非布索坦,或先合成噻唑环,然后引入氰基,水解后得到非布索坦。
2、菲诺贝特[5]
菲诺贝特为第二代苯氧芳酸类药物,可显著降低甘油三酯(TG)、适度降低总胆固醇和低密度脂蛋白胆固醇并能升高高密度脂蛋白胆固醇,有良好的调脂作 用,因而在临床上得到了广泛应用,1998年由雅培公司在美国上市。
菲诺贝特的合成:对羟基苯甲酸用氯化亚砜酰氯化,再与氯苯进行弗克反应,制得4-羟基-4’-氯二苯甲酮,然后再和2-溴-2-甲基异丙酯在碱存在下反应得菲诺贝特。
3、甲磺酸加贝酯[6]
甲磺酸加贝酯是一种非肽类蛋白酶的抑制剂,可抑制胰蛋白酶,激肽释放酶,纤维蛋白溶酶,凝血酶等蛋白酶的活性,从而制止这些酶所造成的病理,用于急性轻型(水肿型)胰腺炎的治疗,也可用于急性出血坏型胰腺炎的辅助治疗。1978年由小野制药公司在日本上市。
甲磺酸加贝酯的合成:6-氨基己酸与S-甲基异硫脲硫酸盐反应,盐酸化得6-胍基己酸盐酸盐 ,与由对羟基苯甲酸乙酯经氯化亚砜缩合得到的4,4’-(亚硫酰二氧基)二苯甲酸二乙酯反应,得加贝酯盐酸盐,再和甲磺酸反应得甲磺酸加贝酯。
4、硝碘酚腈
硝碘酚腈是一种兽药,是一种新型杀肝片吸虫药,能阻断虫体的氧化碳酸化作用,降低ATP浓度,减少细胞分裂所需能量而导致虫体亡。
硝碘酚腈的合成:以对羟基苯甲酸为原料,和尿素、氨基磺酸和对甲酚均匀混合后,升温反应,过滤、洗涤、滤液减压蒸馏,得对羟基苯甲腈[7]。对羟基苯甲腈在冰醋酸中与浓硝酸反应合成出3-硝基-4-羟基苯甲腈,再与碘及过氧化氢在酸性乙醇溶液中反应合成出硝碘酚腈[8]。
5、硝呋齐特[9]
硝呋齐特是一种广谱抗菌药,防治大肠杆菌、沙门氏菌、巴氏杆菌(包括里氏杆菌)、产气杆菌、变形 杆菌、坏杆菌及葡萄球菌等引起的肠道或泌尿系统疾病,可用于疗水产动物的细菌、弧菌及真菌引起的肠道及全身性疾病,防治鸡的球虫病,白细胞原虫引起的白冠病以及盲肠肝炎等病症。
硝呋齐特的合成:对羟基苯甲酸酯化生成对羟基苯甲酸甲酯,和肼反应生成对羟基苯甲酰肼,然后在5-硝基糠醛反应得硝呋齐特。
6、茴拉西坦[10]
茴拉西坦为脑功能改善药,本品对于脑溢血、脑梗、短暂性脑缺血、脑炎以及脑震荡、脑挫伤后的头痛、头晕、肢体麻木、乏力、睡眠困难等脑功能障碍均有改善作用。可用对甲氧基苯甲酸制得对甲氧基苯甲酸酰氯,再和吡咯烷酮缩合而成。
7、胺碘酮[11]
胺碘酮又名乙胺碘呋酮,是属Ⅲ类抗心律失常药,用于用于利多卡因无效的室性心动过速和急诊控制房颤、房扑的心室率。可由对甲氧基苯甲酸酰氯和2-丁基苯并呋喃缩合,再脱甲基、碘化、和二乙胺基氯乙烷缩合而得。
8、对羟基苯甲酸葡萄糖苷酰胺[12]
聂耀,杨巧荷等人合成天麻素的类似物对羟基苯甲酸葡萄糖苷酰胺类化合物,希望能在方面能够获得一些新的信息
四、在农药合成中的合成应用
1、除草剂
1.1、溴苯腈(3,5-二溴代-4-羟基苯甲腈)及其辛酸酯、钠盐、钾盐[13]
溴苯腈及其辛酸酯、钠盐、钾盐是具有一定内吸活性的触杀型除草剂,主要用于小麦、大麦、燕麦、黑麦等谷物,亚麻和非耕作区除草,该药无残留活性。
溴苯腈的制备:对羟基苯甲酸、尿素、氨基磺酸和对甲酚均匀混合后,反应得对羟基苯甲腈。将对羟基苯甲腈、乙醇、水、浓盐酸混合溶解后,升温,和溴反应得溴苯腈。
将辛酰氯和溴苯腈反应制得溴苯腈辛酸酯[14]。溴苯腈和相应的碱制得钠盐、钾盐的水剂。
1.2、碘苯腈(3,5-二碘代-4-羟基苯甲腈)[13]
碘苯腈为触杀型除草剂,主要用于麦类、玉米、高粱等除阔叶杂草。
碘苯腈的制备:先用对羟基苯甲酸制得对羟基苯甲腈,然后对羟基苯甲腈在氯气催化下和碘反应制得。
将辛酰氯和碘苯腈反应制得碘苯腈辛酸酯[15],也有类似的作用。
2、杀虫剂
2.1、杀螟腈[16]
杀螟腈是一种广谱杀虫剂,特别对水稻螟虫、稻苞虫、稻飞虱、稻纵卷叶虫、叶蝉、粘虫等防治效果显著。
杀螟腈的制备:先用对羟基苯甲酸制得对羟基苯甲腈,再和O,O-二甲基硫代磷酰氯反应得到杀螟腈。
2.2、苯腈磷[17]
苯腈磷是一种杀虫剂,对稻螟虫、棉铃虫及鳞翅目幼虫等害虫有效。
苯腈磷的制备:先用对羟基苯甲酸制得对羟基苯甲腈,再和O-乙基苯基硫代磷酰氯反应得到。
五、在染料中的应用
1、紫外线吸收剂
紫外线吸收剂能强烈吸收紫外光,同时也广泛应用的助剂,于日用化工、医药、农药、塑料、涂料等领域,特别实在当前臭氧层破坏严重,太阳紫外线辐射也愈加严重,紫外线吸收剂的应用也越来越受重视。二苯甲酮类衍生物是常用的紫外线吸收剂如2,3,4,4’-四羟基二苯甲酮,4,4’-二羟基二苯甲酮等。
2,3,4,4'-四羟基二苯甲酮的合成[18]:以焦性没食子酸和对羟基苯甲酸为原料合成 2,3,4,4’-四羟基二苯甲酮。
4,4’-二羟基二苯甲酮的合成[19]:以对羟基苯甲酸和苯酚为原料,在氯化锌、三氯氧磷催化下,通过傅克反应合成了4,4'-二羟基二苯甲酮。
2、热敏染料显色剂[20]
1954年美国的NCR(National Cash Register Co Ltd.,现名Apleton Papers Ine)公司首先推出了商品化的压敏、热敏记录,其优异性能,立即引起了广关注,目前已广泛应用于各种领域。而热敏成色剂的的研法生产,也取得了巨大的发展。一般,热敏染料单独存在并不显色,它与显色剂作用下才能形成可逆变色的的热变色混合物,而对羟基苯甲酸苄酯就是常用的热敏染料显色剂。
组合化学是一门将化学合成、组合理论、计算机辅助设计及机械手结合一体,并在短时间内将不同构建模块用巧妙构思,根据组合原理,系统反复连接,从而产生大批的分子多样性群体,形成化合物库(compound library),然后,运用组合原理,以巧妙的手段对库成分进行筛选优化,得到可能的有目标性能的化合物结构的科学。
组合化学与传统合成有显著的不同。传统合成方法每次只合成一个化合物;组合合成用一个构建模块的n个单元与另一个构建模块的n个单元同时进行一步反应,得到n×n个化合物;若进行m步反应,则得到(n×n)m个化合物。有人作过统计,一个化学家用组合化学方法在2~6周的工作量,十个化学家用传统合成方法要花费一年的时间才能完成。所以,组合化学大幅度提高了新化合物的合成和筛选效率,减少了时间和资金的消耗,成为20世纪末化学研究的一个热点。
组合化学的合成技术及对传统药物合成化学的冲击 组合化学合成技术
组合化学合成包括化合物库的制备、库成分的检测及目标化合物的筛选三个步骤。化合物库的制备包括固相合成和液相合成两种技术,一般模块的制备以液相合成为主,而库的建立以固相合成为主。
固相技术 液相技术
优点 纯化简单,过滤即达纯化目的,反应完全;合成方法可实现多设计;操作过程易实现自动化 反应条件成熟,不需调整;无多余步骤;适用范围宽。
缺点 发展不完善;反应中,连接和切链是多余步骤;载体与链接的范围有限 ;反应可能不完全;纯化困难;不易实现自动化。
1多针同步合成
多针同步合成是固相合成的基该方法。将96只带有载体针的小棒固定在同一块板上,其位置与96孔滴度板相对应,然后在96个孔中分别加入不同的反应物及试剂,即可同步合成96个样品。
Dewitt等对此法进行改进,使用下图装置(装置图 动画),在玻璃管的上端加一个硅橡胶垫片,可用注射器加样,管的外面有一个列管式夹层,可对反应物加热或冷却。
他们以此装置合成40个乙酰脲衍生物和具有生理活性的1,4-苯并二氮卓衍生物。(反应式6-43 动画)
2混合-分离随机合成法
1991年Lam等报道了以树脂为载体,进行随机合成,可以同步合成上百万个分子,并提出一珠一肽的概念。首先将19种保护的天然氨基酸分别连在树脂上,混合脱除保护,再分成19份分别与19种保护氨基的氨基酸进行偶联反应,可以得到19×19种连在树脂上的二肽,如此进行五次,可合成出195=
2,476,099种连在树脂上的侧链保护的五肽,脱除侧链保护但不从树脂上切下,可得到由近2.5万连在树
脂上的不同肽段的五肽组成的肽库。此法保证同一树脂上的肽段序列是相同的,即“一珠一肽”。
用该肽库与受体分子反应,可形成显色络合物的肽段树脂就会由无色变为有色,在显微镜下把显色的树脂拣出,用8摩尔/升的盐酸胍洗掉络合物后,用微量多肽测序仪即可测出该肽序列。
Lam等用该法合成的五肽库对抗β-内啡肽的单克隆抗体进行了亲和性研究,找到天然抗原位点肽的六个有效类似物,还用该肽库进行了结合抗生蛋白链菌素的研究,找到一些有结合作用的肽段。
(混分法示意图)
一珠一肽法的优点是可以同步合成大量的化合物,并可对多种受体进行筛选,但只适合于合成能微量测定的样品,如多肽和寡核苷酸,应用范围不广。
3编码确定结构的同步合成
编码确定结构的同步合成法在同步合成时,引入另一个容易合成且在合成后可以通过微量分析测定结构的分子,以该分子作为密码来确定与其同时合成的目标分子的结构。
Mikolaiev 等在1993年报道了Selectide编码合成方法,即在一个树脂上合成一个非肽类化合物或其它不可测序的化合物时,在其上合成一个作为编码用的多肽。
该法常用含多功能团的化合物如Lys等作为目标分子与编码分子的连接点,每一个氨基酸代表目标分子中的一个组成部分,在混合-分离合成法中,每安装一个构建模块,就向目标分子的编码臂上偶联一个代表该构建模块的氨基酸,合成并测定活性后,活性分子结构可以通过测定同一树脂珠上多肽的序列而给出。
药物的开发是一个耗时耗费的过程,据报道,一种新药从开始研制到上市,需8~10年的时间,研究费用高达2~5亿美元。药物的研制历程之所以这样长,很重要的原因是先导化合物的发现与优化速度缓慢。组合化学能够大大加快化合物库的合成及筛选速度,从而大大加快了新药的研制速度。
应用
1新材料的开发
十年来,已报道许多以组合化学方法开发的新材料,如抗磁材料、磷光材料、介电材料、铁电材料、半导体、催化剂、沸石和聚合物及复合材料等。
2催化剂筛选
催化剂传统的筛选法是试凑法,工作量大,效率不高。
科学家们用各种方法设计和建立了催化剂库,对催化剂进行快速筛选,已取得不少成果。
美国Purdue大学开发一种自动制备并检测沸石分子筛的系统,每个式样板有8~19个反应室(150-300微升),每次可同时试验六块板,产品用离心方法回收,最后形成的组合库用X-射线散射技术检测或用电子显微镜筛选,仅消耗很少试剂就取得很多数据。
由于催化反应是放热反应,有活性的催化剂可红外成相。Steven J.Taylor和James P.Morken利用红外热谱仪对载有3000多个潜在催化剂库的聚合物珠进行筛选,找出两个活性有机化合物作为亲核酰化的有效催化剂。
Wilhelm F.Maier 和助手组装了由37种氧化物组成的催化剂库,测定其在100℃对己烯-1氢化的催化活性,红外成相表明有四个点比衬底热,即表明这四个点有活性。活性与非活性点温差非常小,不到0.7℃,但像0.1℃的温差也能可靠地检测。
加州大学Selim M.Senkan教授发展了一基于激光的方法,以快速筛选环己烯脱氢成苯的固相催化剂库,筛选出由80%铂、10%钯和10%铟组成的三元混合物,比库中其他成员生成的苯多。66个成员库使用全自动装置制备,制备和筛选只需两天半时间。
3新药物的合成与筛选
迄今为止,组合化学最多的应用是新药物的设计、合成和筛选方面。R.F.Service在Science撰文,认为组合化学方法创制的新药将冲击21世纪的药物市场。美国及欧洲已涌现一批组合化学公司,杜邦制药公司的研究者将组合化学(随机设计,合理筛选)与合理药物设计(合理设计,随机筛选)两种不同的方法联用设计合成了新奇的胶原酶抑制剂,能够抑制引起癌转移和关节炎的胶原酶(Coll-agenases),这些工作有利于获得更加有效的抑制癌细胞转移和治疗关节炎的新药。
Beatrice Ruhland 以组合化学法,把同手性氨基酸衍生的胺键合到Tenta GelS树脂上,并与非手性烯酮和芳香醛或α,β-不饱和醛发生环加成反应合成了一些3-氨基-2-氮杂环丁酮--制备α-酰胺基-β-内酰胺,包括许多重要的抗生素的前体。发现该反应有很高的cis选择性,二种非对映体cis β-内酰胺比率为1:1到3:1。
1994年,Ellman小组应用多针同步合成系统二次共合成192种结构不同的1,4-苯并二氮杂卓衍生物,并测定了这些化合物对缩胆囊肽(CCK)A受体的结合作用。
Haskell-Luevano,C.等1999年报道以组合化学法固态合成951个化合物,这些化合物用显色生物试验在10μM测试,显示对MC1R分型的活性。选择其中二种重新合成、纯化和鉴定,一种鉴定结构为2的,对鼠的MC1R分型EC50为42.5μM,为进一步研究非肽杂环兴奋剂提供新的起点。
4新农药的合成和筛选
1962年,美国女作家蕾切尔·卡逊撰写《寂静的春天》一书,提出农药野生动物、危害儿童健康、污染表土的问题,引起各国的关注。随后,一批高毒、高残留农药被禁用,并促使农药的研究和生产向提高原药固有的活性及其使用效率和效果,降低农药用量,提高农药对人、畜和作物的安全性,改善与环境的相容性,减少对非靶标生物和生态环境的负面影响的方向发展。
十年来,组合化学法结合高通量的筛选,大大加快农药研究开发的速度,如艾格福公司每年可合成5万个新化合物;诺华公司的筛选能力是每年10万个新化合物;捷利康公司1995~1997年,化合物的筛选能力从每年1万个提高到10万个,1998年为12万个,2000年为20万个。
John J.Parlow 利用分子反应活性的互补性/分子识别技术(CMR/R)平行合成具有除草活性的取代杂环酰胺化合物,生物试验结果表明化合物3有一定的除草活性。(反应式6-46 动画)
他们把3(结构式3)分成两部分。先对A部分的杂环进行改造,改变环上的原子和取代基,得到56个化合物,但生物试验表明它们的活性不如3大;接着对B部分进行改造,以不同的取代基取代苯环C或D的不同部位,得到68个化合物,生物试验表明化合物4(结构式4)的生物活性是3的4倍。
展望
21世纪是绿色化学的世纪。绿色化学要求将原子重新巧妙组合,实现零排放的原子经济反应,生产环境友好产品。所以,组合化学是实现绿色化学的必经之路。
正如中国军事医学研究所胡文祥所长在《广义组合化学》一文所指出的:任何成功的事情或事物都是巧妙的合理的组合。1234567七个音符可以组合成最美妙的音乐旋律,赤、橙、黄、绿、青、蓝、紫七色光可以组合成美丽的画卷和五彩缤纷的世界;喜、怒、哀、乐、悲、恐、惊七种感情可以组合成斑斓的人生。我们相信元素周期表上109种元素的巧妙组合,将为绿色化学、为美化地球环境谱写不朽的篇章。组合化学从一诞生起,便显示出强大的生命力,十余年来,在有机(包括药物)领域得到了蓬勃发展。21世纪的化学将更多地向生命、材料领域渗透,对于这个领域内的合成化学家来说,组合化学提供了一条新的化学合成思路。虽然还面临着诸如缺乏系统有效的平行检测手段等困难,但随着电脑技术和自动化水平的提高及新型检测仪器的研制,这些困难将逐步被解决。
作者:吉民 定价:¥ 35.00 元
出版社:化学工业出版社 出版日期:2004年06月
ISBN:7-5025-5500-5 开本:16 开
类别:有机化学化工 页数:304 页
简介
本书从组合化学的角度出发,详细分析了合成策略,以此为基础着重介绍了固相组合和液相组合的合成方法、组合化学的筛选及低聚物的合成等内容。同时强调了组合化学在高通量筛选和新药发现中的作用,并且对组合化学的进展做了展望。
目录
第1章组合合成策略7
混合?裂分法7
树脂珠技术9
茶叶袋法10
平行合成法10
使用树脂珠的反应器械14
多中心合成法15
空间定位平行合成法15
混合试剂合成法16
参考文献16
第2章组合合成方法——固相组合合成18
载体19
树脂珠19
多针22
圆片24
薄片25
结合分子28
酸不稳定结合分子31
碱不稳定结合分子34
安全制动结合分子37
氨基甲酸酯结合分子38
硅结合分子/无痕迹结合分子40
光不稳定结合分子41
烯丙基官能团化结合分子42
多处可裂(多官能团)结合分子42
多中心结构库模板43
方酸44
经Baylis?Hillmann反应得到的模板45
2?溴乙酰基)吡咯作为模板51
用烯酮作为模板54
反应类型59
亲电和亲核取代反应60
取代反应63
杂环合成63
环加成反应66
缩合反应67
酰胺形成及相关反应69
及相关反应69
麦克尔加成69
烯烃形成69
氧化反应69
还原反应69
参考文献70
第3章组合合成方法——液相组合合成76
与固相组合合成相比较77
混合物的合成78
已用于液相组合化学的反应80
酰化反应80
胺的磺化80
脲、硫脲和氨基甲酸酯的制备81
烷化和加成反应81
还原胺化81
胺的芳基化81
经缩合反应形成碳?碳键81
钯催化的碳?碳键的形成81
氢化和还原81
多组分反应81
环化反应82
其他反应82
反应顺序83
纯化85
固相束缚试剂85
固相萃取86
液相萃取88
氟的合成89
在可溶性聚合物载体上的合成91
6树枝状载体93
高聚物试剂的使用94
参考文献95
第4章组合化学库的筛选97
混合物库97
在珠筛选法97
解缠绕法98
编码105
多处可裂的结合分子115
含单独化合物的库115
参考文献116
第5章组合化合物库的鉴定118
红外光谱法(IR)118
傅里叶?红外显微镜学118
衰减全反射光谱127
其他的红外光谱方法129
核磁共振法130
在珠分析法130
高分辨质子魔角旋转核磁共振133
3质谱138
组合化合物分析138
样品分析与纯化的高通量系统154
参考文献160
第6章组合合成的低聚物165
6?1类肽165
亚单体法165
单体合成法167
3拟肽物167
彻底烷基化多肽168
类肽169
低聚氨基甲酸酯169
磺酰肽和插烯磺酰氨肽170
聚?N?酰胺172
寡脲174
线性寡脲174
低聚环脲和环硫脲174
硫脲175
脲类肽175
含杂环低聚物175
聚甲基吡咯和咪唑175
含噻唑环和?唑环的多肽176
寡聚四氢呋喃176
聚异?唑啉177
寡聚噻吩177
含吡咯啉酮的低聚物177
其他合成低聚物178
反假肽178
插烯多肽179
氮杂化物和氮杂多肽179
多肽180
四取代氨基酸的多肽180
聚羟基化合物181
多肽核酸181
肽键在一个位置上的修饰182
硫代酰胺假肽182
酰胺键被还原的多肽182
羟基酰胺键的多肽183
羟乙胺肽键电子等排体184
参考文献184
第7章自动组合合成188
单个化合物的平行合成189
实验室制备效率189
实验室自动化设备190
分散型自动化系统191
中心控制和功能型多组分系统192
中心自动的样品导向多组分系统192
高通量纯化和分析193
自动纯化193
微反应系统简介194
参考文献195
第8章组合生物合成196
克隆生物合成功能基因簇196
遗传工程及新药研究197
1靶向基因失活197
单基因表达198
基因簇的表达202
合成起始单位变异203
酶亚基的重组装203
组合生物合成的应用212
寡糖类抗生素生物合成基因的运用212
其他来源基因的运用——地球上的新化合物212
对酶变换其底物特异性212
参考文献213
第9章用作化学传感器的分子接受器215
超分子识别部位215
大环肽类217
组合接受器库218
环肽作为化学传感器的超分子识别部位219
参考文献222
第10章高通量筛选与新药发现224
高通量药物筛选224
对高通量筛选的要求225
高通量药物筛选的组成226
化合物资源226
微反应系统227
筛选模型227
高灵敏度检测系统229
自动化操作系统230
数据采集传输处理系统231
高通量筛选的特点231
高通量药物筛选的过程232
高通量筛选系统简介233
虚拟筛选234
参考文献236
第11章催化反应的高通量实验238
1HTE技术用于催化反应238
库设计和试验策略240
合成方法242
测试方法243
多路径反应器244
参考文献246
第12章计算机辅助化合物库设计247
化合物库设计理论248
相似性原则249
分子描述250
二维指纹250
三点药效团251
其他描述251
分子相似性方法252
亲和力指纹252
特征树253
碎片的自动化结构重合254
描述有效性研究254
和3D描述的对比254
随机设计和合理化设计的比较255
三点药效团和2D指纹比较256
局部相似——相似性半径256
化合物选择技巧257
设计组合化合物库258
参考文献262
第13章组合化学进展264
丝氨酸及半胱氨酸蛋白酶抑制剂265
真菌I型蛋白香叶基转移酶(GGTase?1)抑制剂267
KDR受体酪氨酸激酶抑制剂268
自动形成靶向化合物库设计270
优先GPCR配体272
拮抗剂274
胺的合成277
多样性导向合成280
Katritsky苯并三唑固相合成法283
多组分缩合285
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。