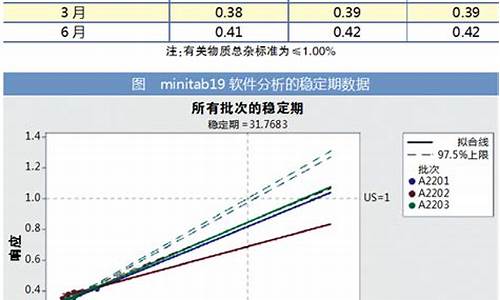
原料药结晶-原料药晶型对含量影响有哪些

摘要 本文就如何建立TLC法测定有关物质的方法进行论述,系统地阐述了薄层色谱法各条件确定的原理,并列举了质量标准制订中存在的某些问题。
关键词 薄层色谱法(TLC法) 有关物质 方法建立
有关物质是研究药品中除主成分以外的杂质,它可能是原料药合成过程中带入的原料、中间体、试剂、降解物、副产物、聚合体、异构体以及不同晶型、旋光异构的物质,也可能是制剂过程中产生的降解物,或是在贮藏、运输、使用过程中产生的降解物等[1]。这些杂质的存在直接反映药品的有效性和安全性,故要对其进行研究,特别是在药品申报的质量研究资料中需建立其检测方法,并根据生产、稳定性考核等实际情况考虑是否在质量标准中制订该检查项,规定其限度。目前,有关物质的常用测定方法有高效液相色谱法(HPLC法)和薄层色谱法(TLC法)。
TLC的特点是快速、简便,尤其是对无紫外吸收的杂质测定,更具有其应用价值。如能将TLC法与HPLC法有机地结合、或彼此间进行比对研究,便可得到更多、更为准确的有关杂质信息,做到两方法间的相辅相成,相益得彰!本文将着重讨论如何建立薄层色谱法测定有关物质的方法。
1.测定方法类型
常用的方法有杂质对照品法(适用于已知杂质)和自身(稀释)对照法(适用于一般杂质检查,杂质成分少且尚不能取得杂质对照品)。目前国内由于难以获得杂质对照品、故一般均采用自身对照法。
2.展开剂的确定(即专属性试验)
专属性的研究是提供被分析物在杂质和辅料存在时能被区分的证明,该点是色谱条件建立的关键。通常采用在被分析物的对照品或精制品中加入一定量的杂质或辅料,证明色谱条件可将各杂质与被分析物分离[1]。这里的关键是:将多少量的杂质加入到多少量的主成分中。正确的作法是将1%(w/w)浓度量的各杂质加入到100%浓度的主成分中,配制这样的溶液来
验证系统适用性。之所以如此配制,目的是模仿样品中有可能存在的状态,即有少量(1%左右)杂质存在时是否能与主成分达到完全分离,只有这样才能比较客观、科学地反映样品中实际存在情况的(见图1);而不应把该溶液配制成:主成分与中间体相同浓度的。因为一者实际检测时样品中不可能存在此种情况;二者该浓度不易确定,目前国内申报资料中一般的作法均是配制成较低的一致浓度,这样各斑点当然易于完全分离了(见图2),但在实际测定时,由于主斑点急剧增大,很易将相邻杂质包含于主成分斑点中。同样,质量标准中的系统适用性试验用溶液的配制方法亦如此。
(1,3,4为杂质,2为主成分)
图1 图2 (杂质浓度均为供试品溶液浓度的1%)
3.检出条件的确定
其基本出发点是:主成分与相关杂质均应在该条件下显色,且在相同浓度下,斑点大小应基本一致。薄层板的类型根据被测物质的性质来选用,测定有紫外吸收的物质通常选用GF254或GF365板;测定无紫外吸收、需喷显色剂的,常选用硅胶G板或H板,选用该类薄层板时,显色方法根据被测物质的结构式选取,但当有多个显色方法时,应分别进行试验,选取灵敏度最高的显色方法。如醋酸氢化可的松有关物质的测定,中国药典2000年版采用碱性四氮唑蓝试液显色,美国药典26版采用硫酸-乙醇(10:90)溶液显色,两者均为激素类药物的显色方法。醋酸氢化可的松属于激素类中的肾上腺皮质激素,四氮唑法是肾上腺皮质激素的重要显色方法;而硫酸-乙醇显色法则主要是针对激素类中的雌激素的显色反应,对于属于肾上腺皮质激素类的醋酸氢化可的松则反应活性不强,结果两法的灵敏度相差10倍以上。因此,检出条件的确定,一定要在查阅文献的基础上,并根据试验结果进行综合考虑。
4.供试品溶液浓度的确定(灵敏度试验——最低检出限的测定)
供试品溶液浓度的设定在有关物质检测中是至关重要的,浓度越高、越能反映样品中杂质存在的情况,但若设定得过高,则会产生主斑点严重拖尾、“断腰”等超载现象的发生,产生错误结论;若设定太低,又将达不到检测杂质的目的,观测不到杂质量的变化。其设定是根据最低点样量和最大点样量来综合考虑的。
最低检出限虽然是个绝对值,但真正的意义却是其相对值,即相对于供试品溶液的浓度多少而言,所以测定结果不仅要罗列出其绝对值又应列出其相对值,这样最低检出限才有意义!最大点样量则是通过不断加大供试品溶液浓度,直至主斑点严重拖尾、“断腰”等情况出现时来得到的。然后根据最低检出限,采用“上推法”来确定:如一般设定杂质斑点小于1.0%对照斑点,对照溶液的浓度至少应为最低检出浓度(即最低检出限)的20~50倍,则供试品溶液浓度是最低检出浓度的2000~5000倍;反过来,最低检出浓度应至少达到供试品溶液浓度的0.02%~0.05%。应注意的是:由于最低检出量和最大点样量因试验环境、薄层板质量以及即时试验时其他各因素的不同而改变(即耐受性因数),故供试品溶液的浓度在保证小于最大进样量的情况下,可在此基础上设定得再高一些,以保证该浓度可适用于各种条件下。举例说明见表1(规定杂质限度为1.0%)。
表1 最低检出量、最大点样量、供试品溶液和对照溶液浓度之间的比例关系
最大点样量
供试品溶液
对照溶液
最低检出量 浓 度 8mg/ml 3mg/ml 30μg/ml 1μg/ml 点样量 10μl 10μl 10μl 10μl 绝对量 30μg 0.3μg 10ng 相对于样品测定浓度的 100% 1.0% 0.02% 倍 数 关 系 5000倍 30倍 “基准点”
供试品溶液浓度也可设定得再高些,但不可超过最大点样量。
5.加样回收试验(即准确性试验)
准确性试验可采用在预经有关物质测定后的样品中,加入已知量杂质的方法来评价。准确称取各杂质,将含有1%(w/w)浓度的各杂质加入到样品溶液中,以验证所采用的薄层测定条件是否可分离检测出相应的各杂质以及样品中已存在的杂质是否累加,斑点是否加深。该原理同前面所述的专属性研究是一致的。
6.强力破坏试验
该项研究是为了揭示原料药内在稳定性的特性,它是开发研究的一部分。这些试验是在比加速试验更剧烈的条件下进行的,其能够包含药品在销售过程中所遇到的剧烈条件。可取一批样品通过强光、高温、高湿、氧化破坏、以及酸碱破坏来证明该展开条件能分离检测出杂质。
7.展开距离
测定时一定要采用25cm、长薄层板,展开距离应尽可能长一些,以使杂质与主成分尽量分离。如用短板,易造成临近主斑点的杂质斑点“躲进”主斑点中。但同时又应注意,距离拉大,斑点分散,会损失最低检出限,降低灵敏度,故应综合考虑。
8.其它的因素
展开温度应尽量控制在20~25℃之间,尤其在冬季,应注意环境的温度,如太低,将严重影响展开效果。另层析缸的盖儿,应涂抹凡士林油,以保证整个试验过程中,层析缸的密封,避免展开剂挥发;并应在展开前,预先倾入展开剂,以使层析缸内的空气饱和,达到最佳的展开效果。薄层板由于有自制、市售,质量不一也应注意。
二.讨论
1. 质量标准中的系统适用性试验,最好能将最难分离的杂质订入系统适用性试验用溶液的配制,将此杂质的浓度配制为主成分浓度的1%,或0.5%,或0.2%(依据杂质限度而定)进行试验,验证分离度后,再进行样品的测定,以确保试验的准确进行。
2. 质量标准中,应配制系列浓度的对照溶液(即梯度对照),以对杂质有“半定量”的概念,这可更好地评价杂质存在的情况;并应规定杂质的个数及最大杂质斑点的限度,使质量标准更完善、科学。经查阅,中国药典薄层色谱法测定有关物质的有70个品种,仅有2个品种采用了梯度对照,绝大部分品种仅是制定了对照溶液,均未规定杂质个数,和最大杂质斑点限度,如有若干个杂质斑点也无法判定;而英国药典和美国药典则几乎每个品种均采用梯度对照,并规定杂质个数和最大杂质斑点限度,这一点值得学习和推广。
3. 错误的一种误区,认为HPLC法完全替代了TLC法,这是不正确的,一定要做到相互补充、相互论证、相互参考才是最客观、最科学的!
本文是在参阅了日本《分析方法验证》一书和大量日本国内新药申报资料中质量研究部
分的内容所写而成。
如何建立薄层色谱法测定有关物质的方法
化学药品在没制作成制剂(方便有利于服用使用的形式)成品前叫化学原料药,原料药的原粉是一种化学纯净物资,每一种纯净的化学物资都有它自己的独特的物理和化学性质。表征药品的性质的常规项目有外观、熔点、和含量。熔点是特定的,每种药品都不同。药品还有特殊的性质,就是生物活性,生物活性与药品的结晶型和旋光性有关,结晶型和旋光与物质在介质中温度和介质有关。红外线是含有能量的光波,照射在药品上达到一定温度就会改变药品晶型或熔融表面,破坏他的晶型或旋光,改变他的生物活性,也许就生成了有副作用的物质。还有一些生物制剂药和化学不稳定药都是怕光的。这样的解释您是否满意
撒隆巴斯进口和国产区别
摘要 本文就如何建立TLC法测定有关物质的方法进行论述,系统地阐述了薄层色谱法各条件确定的原理,并列举了质量标准制订中存在的某些问题。
关键词 薄层色谱法(TLC法) 有关物质 方法建立
有关物质是研究药品中除主成分以外的杂质,它可能是原料药合成过程中带入的原料、中间体、试剂、降解物、副产物、聚合体、异构体以及不同晶型、旋光异构的物质,也可能是制剂过程中产生的降解物,或是在贮藏、运输、使用过程中产生的降解物等[1]。这些杂质的存在直接反映药品的有效性和安全性,故要对其进行研究,特别是在药品申报的质量研究资料中需建立其检测方法,并根据生产、稳定性考核等实际情况考虑是否在质量标准中制订该检查项,规定其限度。目前,有关物质的常用测定方法有高效液相色谱法(HPLC法)和薄层色谱法(TLC法)。
TLC的特点是快速、简便,尤其是对无紫外吸收的杂质测定,更具有其应用价值。如能将TLC法与HPLC法有机地结合、或彼此间进行比对研究,便可得到更多、更为准确的有关杂质信息,做到两方法间的相辅相成,相益得彰!本文将着重讨论如何建立薄层色谱法测定有关物质的方法。
1.测定方法类型
常用的方法有杂质对照品法(适用于已知杂质)和自身(稀释)对照法(适用于一般杂质检查,杂质成分少且尚不能取得杂质对照品)。目前国内由于难以获得杂质对照品、故一般均采用自身对照法。
2.展开剂的确定(即专属性试验)
专属性的研究是提供被分析物在杂质和辅料存在时能被区分的证明,该点是色谱条件建立的关键。通常采用在被分析物的对照品或精制品中加入一定量的杂质或辅料,证明色谱条件可将各杂质与被分析物分离[1]。这里的关键是:将多少量的杂质加入到多少量的主成分中。正确的作法是将1%(w/w)浓度量的各杂质加入到100%浓度的主成分中,配制这样的溶液来
验证系统适用性。之所以如此配制,目的是模仿样品中有可能存在的状态,即有少量(1%左右)杂质存在时是否能与主成分达到完全分离,只有这样才能比较客观、科学地反映样品中实际存在情况的(见图1);而不应把该溶液配制成:主成分与中间体相同浓度的。因为一者实际检测时样品中不可能存在此种情况;二者该浓度不易确定,目前国内申报资料中一般的作法均是配制成较低的一致浓度,这样各斑点当然易于完全分离了(见图2),但在实际测定时,由于主斑点急剧增大,很易将相邻杂质包含于主成分斑点中。同样,质量标准中的系统适用性试验用溶液的配制方法亦如此。
(1,3,4为杂质,2为主成分)
图1 图2 (杂质浓度均为供试品溶液浓度的1%)
3.检出条件的确定
其基本出发点是:主成分与相关杂质均应在该条件下显色,且在相同浓度下,斑点大小应基本一致。薄层板的类型根据被测物质的性质来选用,测定有紫外吸收的物质通常选用GF254或GF365板;测定无紫外吸收、需喷显色剂的,常选用硅胶G板或H板,选用该类薄层板时,显色方法根据被测物质的结构式选取,但当有多个显色方法时,应分别进行试验,选取灵敏度最高的显色方法。如醋酸氢化可的松有关物质的测定,中国药典2000年版采用碱性四氮唑蓝试液显色,美国药典26版采用硫酸-乙醇(10:90)溶液显色,两者均为激素类药物的显色方法。醋酸氢化可的松属于激素类中的肾上腺皮质激素,四氮唑法是肾上腺皮质激素的重要显色方法;而硫酸-乙醇显色法则主要是针对激素类中的雌激素的显色反应,对于属于肾上腺皮质激素类的醋酸氢化可的松则反应活性不强,结果两法的灵敏度相差10倍以上。因此,检出条件的确定,一定要在查阅文献的基础上,并根据试验结果进行综合考虑。
4.供试品溶液浓度的确定(灵敏度试验——最低检出限的测定)
供试品溶液浓度的设定在有关物质检测中是至关重要的,浓度越高、越能反映样品中杂质存在的情况,但若设定得过高,则会产生主斑点严重拖尾、“断腰”等超载现象的发生,产生错误结论;若设定太低,又将达不到检测杂质的目的,观测不到杂质量的变化。其设定是根据最低点样量和最大点样量来综合考虑的。
最低检出限虽然是个绝对值,但真正的意义却是其相对值,即相对于供试品溶液的浓度多少而言,所以测定结果不仅要罗列出其绝对值又应列出其相对值,这样最低检出限才有意义!最大点样量则是通过不断加大供试品溶液浓度,直至主斑点严重拖尾、“断腰”等情况出现时来得到的。然后根据最低检出限,采用“上推法”来确定:如一般设定杂质斑点小于1.0%对照斑点,对照溶液的浓度至少应为最低检出浓度(即最低检出限)的20~50倍,则供试品溶液浓度是最低检出浓度的2000~5000倍;反过来,最低检出浓度应至少达到供试品溶液浓度的0.02%~0.05%。应注意的是:由于最低检出量和最大点样量因试验环境、薄层板质量以及即时试验时其他各因素的不同而改变(即耐受性因数),故供试品溶液的浓度在保证小于最大进样量的情况下,可在此基础上设定得再高一些,以保证该浓度可适用于各种条件下。举例说明见表1(规定杂质限度为1.0%)。
表1 最低检出量、最大点样量、供试品溶液和对照溶液浓度之间的比例关系
最大点样量
供试品溶液
对照溶液
最低检出量 浓 度 8mg/ml 3mg/ml 30μg/ml 1μg/ml 点样量 10μl 10μl 10μl 10μl 绝对量 30μg 0.3μg 10ng 相对于样品测定浓度的 100% 1.0% 0.02% 倍 数 关 系 5000倍 30倍 “基准点”
供试品溶液浓度也可设定得再高些,但不可超过最大点样量。
5.加样回收试验(即准确性试验)
准确性试验可采用在预经有关物质测定后的样品中,加入已知量杂质的方法来评价。准确称取各杂质,将含有1%(w/w)浓度的各杂质加入到样品溶液中,以验证所采用的薄层测定条件是否可分离检测出相应的各杂质以及样品中已存在的杂质是否累加,斑点是否加深。该原理同前面所述的专属性研究是一致的。
6.强力破坏试验
该项研究是为了揭示原料药内在稳定性的特性,它是开发研究的一部分。这些试验是在比加速试验更剧烈的条件下进行的,其能够包含药品在销售过程中所遇到的剧烈条件。可取一批样品通过强光、高温、高湿、氧化破坏、以及酸碱破坏来证明该展开条件能分离检测出杂质。
7.展开距离
测定时一定要采用25cm、长薄层板,展开距离应尽可能长一些,以使杂质与主成分尽量分离。如用短板,易造成临近主斑点的杂质斑点“躲进”主斑点中。但同时又应注意,距离拉大,斑点分散,会损失最低检出限,降低灵敏度,故应综合考虑。
8.其它的因素
展开温度应尽量控制在20~25℃之间,尤其在冬季,应注意环境的温度,如太低,将严重影响展开效果。另层析缸的盖儿,应涂抹凡士林油,以保证整个试验过程中,层析缸的密封,避免展开剂挥发;并应在展开前,预先倾入展开剂,以使层析缸内的空气饱和,达到最佳的展开效果。薄层板由于有自制、市售,质量不一也应注意。
二.讨论
1. 质量标准中的系统适用性试验,最好能将最难分离的杂质订入系统适用性试验用溶液的配制,将此杂质的浓度配制为主成分浓度的1%,或0.5%,或0.2%(依据杂质限度而定)进行试验,验证分离度后,再进行样品的测定,以确保试验的准确进行。
2. 质量标准中,应配制系列浓度的对照溶液(即梯度对照),以对杂质有“半定量”的概念,这可更好地评价杂质存在的情况;并应规定杂质的个数及最大杂质斑点的限度,使质量标准更完善、科学。经查阅,中国药典薄层色谱法测定有关物质的有70个品种,仅有2个品种采用了梯度对照,绝大部分品种仅是制定了对照溶液,均未规定杂质个数,和最大杂质斑点限度,如有若干个杂质斑点也无法判定;而英国药典和美国药典则几乎每个品种均采用梯度对照,并规定杂质个数和最大杂质斑点限度,这一点值得学习和推广。
3. 错误的一种误区,认为HPLC法完全替代了TLC法,这是不正确的,一定要做到相互补充、相互论证、相互参考才是最客观、最科学的!
本文是在参阅了日本《分析方法验证》一书和大量日本国内新药申报资料中质量研究部
分的内容所写而成。
有毒化学药品溅在皮肤上蚀可用乙醇等有机溶剂擦洗是对还是错
撒隆巴斯进口和国产的区别有价格不同、质量标准不同和辅料质量、原料药不同。
1、价格不同:由于进口药物的成本和关税占据很大一部分,所以药物费用也比较高。
2、质量标准不同:现今的国产药和进口药已经没有明显的区别,部分药物还以国产的质量最好。不过也有少数药物,进口药物和国产药物,因为生产工艺不同,效果上存在一定差异。
3、辅料质量、原料药不同:原料药晶型不同、纯度不同,其溶解度、稳定性不同。国产与进口没有绝对的孰好孰坏,主要取决于制药工艺,比如有些头孢类药物因无法严格控制杂质含量需要做皮试,而制作工艺较好的药物则不需要做皮试。
2018年初级药师考试考点:药物制剂的稳定性
错。直接用乙醇等,会使污染皮肤的面积扩大。
“化学药品”这个词,可能会令大多数人都想到我们在生病时吃的某种西药,这一理解的歧义,主要来自于“药品”二字。其实啊,“药品”并不仅仅是指吃的药物,也包含了化学试剂。所以,我们这里说的“化学药品”,其实指的是化学试剂——做化学实验用的化学物质。
同品种已经药典会核定,统一为核定的名称。
无同品种经药典会核定,要求申报单位与药典会接洽进行通用名称核定。
结构确证
1、说明结构确证测试样品的来源(精制)和纯度。
2、对照品的来源:是否合法。
3、对含多个手性中心的原料药需确定本品的立体结构,长要求补充进行NOE谱 或其他图谱的测定,或提供本品详细的同样测试条件下的核磁共振文献图谱和数据以进一步确定本品的立体结构。
4、晶型研究发补原则。对于难溶性化合物,制剂为口服固体制剂,同时从文献报道已知晶型对生物利用度或稳定性有明显影响,这种情况应在临床研究前要求进行晶型研究,其它情况可不要求。
以上内容来自 百度百科-化学药品
大家做好准备迎接考试了吗?诚意整理“2018年初级药师考试考点:药物制剂的稳定性”,只要付出了辛勤的劳动,总会有丰硕的收获!欢迎广大考生前来学习。
2018年初级药师考试考点:药物制剂的稳定性
一、概述
(一)研究药物制剂稳定性的目的和意义
稳定性是评价药物制剂质量的重要指标之一,也是确定药物制剂使用期限的主要依据。
药物制剂稳定性一般包括化学、物理和生物学三个方面。
(二)药物稳定性的化学动力学基础
化学动力学基础研究药物的降解速度与浓度的关系可用式(12-1)表示。
(12-1)
式中,dC/dt是降解速度;k是反应速度常数;C是反应物浓度;n是反应级数。
n=0为零级反应;n=1为一级反应;n=2为二级反应,反应级数是用来阐明反应物浓度对反应速度影响的大小。
(1)零级反应:反应速度与反应物浓度无关
(2)一级反应:反应速度与反应物浓度的一次方成正比,其浓度与时间的关系为:
(12-3)
(三)温度对反应速率的影响
反应温度越高,药物的降解速度也就越快。
(四)药物稳定性的预测
分解10%所需的时间(即t0.9)或(有效期)。
二、制剂中药物的化学降解途径
药物由于化学结构的不同,其降解反应也不一样,水解和氧化是药物降解的两个主要途径。其他如异构化、聚合、脱羧等反应,在某些药物中也有发生。有时一种药物还可能同时产生两种或两种以上的反应。
1.水解
(1)酯类(包括内酯)
(2)酰胺类(包括内酰胺)
2.氧化
(1)酚类
(2)烯醇类
三、影响药物制剂降解的因素及稳定化方法
(一)影响药物制剂降解的处方因素和稳定化方法
1.pH值的影响
2.广义酸碱催化
3.溶剂的影响
4.离子强度的影响
5.加入表面活性剂
6.处方中赋形剂和附加剂
(二)影响药物制剂降解的环境因素和稳定化方法
1.温度
2.光线
3.空气(氧)的影响
(3)防止氧化的措施
1)配液时使用新鲜煮沸放冷的注射用水。
2)在溶液中和容器空间通入惰性气体如二氧化碳或氮气
3)加入抗氧剂
4.金属离子的影响
可加入螯合剂
5.湿度和水分的影响
6.包装材料的影响
(三)药物制剂稳定化的其他方法
1.改进剂型与生产工艺
(1)制成固体制剂
(2)制成微囊或包合物
(3)采用粉末直接压片或干法制粒压片
(4)采用包衣工艺
2.制成稳定的衍生物
制成难溶性盐、酯类、酰胺类或高熔点衍生物,可增加其稳定性。
引入另一种载体基团(或与另一母体药物结合)形成一种新的化合物,这种化合物在体内经生物转化,释放出母体药物而呈现疗效。
四、固体药物制剂的稳定性
1.固体药物制剂稳定性的一般性特点
2.固体药剂的晶型变化与稳定性的关系
同一药物的不同晶型简单分为稳定型和亚稳型。稳定型熔点高,化学稳定性好,但溶解度和溶出速度较低;亚稳型熔点和化学稳定性较低,但溶解度和溶出速度较高。无定型的分子间力更弱,常有较低的熔点、密度和硬度,更高的溶解度和溶出速度。
3.固体药剂的吸湿
具有水溶性的药物粉末在相对较低湿度环境时一般吸湿量较少,但当相对湿度提高到某一值时,吸湿量急剧增加,此时的相对湿度称为临界相对湿度(CRH)。CRH越小越易吸湿,反之,则不易吸湿。(控制生产)
几种水溶物混合后,其吸湿性有如下特点:“混合物的CRH约等于各药物CRH的乘积,与各组分的比例无关。水不溶物的吸湿性在相对湿度变化时,具有加和性。
五、药物稳定性的试验方法
稳定性试验的目的是考察原料药或药物制剂在温度、湿度、光线的影响下随时间变化的规律,为药品的生产、包装、贮存、运输条件提供了科学依据,同时通过试验建立药品的有效期。
(一)影响因素试验
影响因素试验包括强光照射试验、高温试验和高湿度试验。
pH值与氧及其他条件对药物稳定性的影响,并研究分解产物的分析方法。
(二)加速试验
1.常规试验法
此项试验是在温度(40±2)℃、相对湿度(75±5)%的条件下放置6个月。
2.经典恒温法
预测药物有效期,经常采用经典恒温法。Arrhenius指数定律:
(12-8)
其对数形式为:
(12-9)
式中:k:降解反应的速度常数;E:药物的活化能;R:气体常数;T:绝对温度;A:频率因子。
设计实验温度,求对应的反应速度常数,做曲线,找出相应温度的K值,代入下列公式,求t
(12-10)
(三)长期试验
1.实验方法
长期试验是在接近药品的实际贮存条件(温度(25±2)℃,相对湿度(60±10)%)下放置12个月,其目的为制定药物的有效期提供依据。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。