原料药的含量限度是指-原料药的含量如规定上限为100以上时

含量90%~105%是相对于标样来的说的
比如你HPLC外标法法测含量,你的标样含量为99%,你实际产品的含量为99.5%,你的外标法计算出的含量就大于100%了,反之,你的实际产品含量为98%,你外标法计算就小于100%。
原料药的含量测定结果按什么表示
药品生产疑难解答800问汇总
1 化工产品如氢氧化钠、亚硫酸氢钠按药典标准检验合格,但非药用级是否即可以投生产,如果不可以,做为铺料需要办什么手续吗?如果送省所检验,是否只要送一批,还是批批送检?另外设备、方法、产品生产工艺等不变的情况下,清洁验证是否只需做一次就够了,还是每年都要做呢?多谢.
答:原料药生产使用的原材料相对宽松,但制剂的原辅料已经在药典上有的品种,必须是药用级别或有批准文号的产品,没有上药典的产品可使用食用级别,但要有产品质量标准.其它产品按药典规定执行.任何的验证都必须做三次才有统计学意义.
2 我公司现正准备申报膏剂车间GMP认证,按照《药品生产质量管理规范认证管理办法》申请与审查中(三)药品生产管理和质量管理自查情况(包括企业概况及历史沿革情况、生产和质量管理情况,证书期满重新认证企业软、硬件条件的变化情况,前次认证不合格项目的改正情况)的要求.是不是说自查内容中一定要有“前次认证不合格项目的改正情况”?
答:对,必须有前次,包括上次认证以来所有药监部门的检查整改情况.
3 我们现在需要设计喷雾剂车间,喷雾剂车间是属于非无菌制剂吗?生产环境的洁净级别应该是1万?还是10万?
答:喷雾剂通常属于非无菌制剂,如果是外用药用药,用于非创伤皮肤使用,30万级即可,如果是用于创伤皮肤或口腔给药,则需要十万级以上.
4 统一换发的药品生产许可证,是不是生产范围中原写有的剂型没有通GMP认证,在这次换证时生产范围中就会删除其没有通过认证的范围?(例如:原证写有大容量,小容量,口服剂,冻干粉.而现通过GMP证的只有小容量和口服剂,那此次换证时生产范围内是不是只写小容量和口服剂吗?)
答:大容量注射剂如果是旧范围,将按规定取消范围,新增范围则予保留.
5 对于回顾性再验证有没有次数及时间的限定?
答:没有次数限制,但有时间要求,通常再验证规定为一年,对风险比较在的设备,如空调系统的高效过滤通常定为半年.
6 GMP规定,应该按照处方量的100%投料.对此应该如何理解?这里的100%投料是指:1、按照原料的主药的含量、水分、折算后,每次换算成100%投料?即:如果,我的原料含量为99%的话,我投料应该是:处方量/99%吗?此种情况每次要变具体的投料重量.2、符合原料药质量标准的原料,都按100%处理,都投处方量.此种情况,每次投料量固定.请问,哪种理解应该更准确呢?
答:第一个操作比较适合生产要求,因为生产中的产品规格通常是含量规格,如片剂,则以一片药品的主药含量为规格,用换算后的100%理论需要原料投料,生产出来的产品规格才符合要求.生产的药品具有本品原有的均一性、含量和效价、质量和纯度.按处方配制的药品,保证其活性成份含量不低于100%标示量或规定量.
7 GMP规定,应该按照处方量的100%投料.对此应该如何理解?这里的100%投料是指:1、按照原料的主药的含量、水分、折算后,每次换算成100%投料?即:如果,我的原料含量为99%的话,我投料应该是:处方量/99%吗?此种情况每次要变具体的投料重量.2、符合原料药质量标准的原料,都按100%处理,都投处方量.此种情况,每次投料量固定.哪种理解应该更准确呢
答:第一个操作比较适合生产要求,因为生产中的产品规格通常是含量规格,如片剂,则以一片药品的主药含量为规格,用换算后的100%理论需要原料投料,生产出来的产品规格才符合要求.生产的药品具有本品原有的均一性、含量和效价、质量和纯度.按处方配制的药品,保证其活性成份含量不低于100%标示量或规定量.
8 无菌原料药用铝桶是否可以重复使用?为什么?
答:不可以重复使用.回收的产品不符合GMP要求,另一方面对无菌产品存在比较大的质量管理风险.
9 除标签、说明书外如何界定按标签类管理的物料?只有品名、规格、商标而没有用法用量等内容的包材(如大箱、铝箔)可否不按标签类物料管理?
答:小盒也属于按标签管理的物料.大箱、无印刷的铝箔按普通物料管理即可.
10 批生产记录是不是一定要有一定总的批生产指令,我公司实际情况无法做到一开始就给定一个总的批号,批包装记录时就有一个总的批包装指令.这种情况,实际生产要如何做,是每个工序下生产指令还是象传统先给定一个批号再进行生产,或者只要将该最终批号的相关工序记录整理一块和批包装记录装订起来成一个批的批生产记录?
你公司的操作与GMP要求相左,不符合要求.没有批生产指令就不能生产,没有生产批号不能有批指令,这样不符合规范.需要继续学习法规及培训.
11 中药厂对购回的中药材质管部自已经过鉴定后可作为标本吗?负责中药标本鉴定的人必须具备哪些资格,我公司员工具有中药专业本科学历,但职称是工程师,他具体中药标本鉴别的资格吗?
答:对于企业内部使用的标本,目前法规上没有明确的规定,有经验的老药工、中药学的工程技术人员都可认为适合.
12 境外公司(美国)在中国投资建药厂,除需要通过美国fda认证外,是否需要通过我国的GMP认证(建成后其所生产的药品全部销往美国,不在中国销售使用)!
答:领取我国《药品生产许可证》的药品生产企业必须通过中国的GMP认证,对于以中间体出去的原料厂则由企业根据情况来决定.
13 建设厂外车间,提出变更生产许可证的时间,应该在申请厂房验收前,还是厂房验收后,还是做替代料生产前申请.
答:申请验收时同时提出申请,验收合格后由药监部门依法进行变更.
14 我公司采购了一批原料,检验成分及含量符合国家标准,但内\外包装标签没有打产品批准文号,经查该厂确实生产了此批原料,请问此原料该怎样处理
答:你厂的供应商考核标准及验货制度是怎样制定的?如果不符合规定可按退货处理.
15 有关药品生产日期和批号确定问题,国家一直没有指导意见,企业做法多种多样,生产日期有采用投料日期或中间日期或成品日期,批号有采用投料日期或中间日期或成品日期或流水号.我个人意见是,因为产品有效期的确定是在产品注册时从成品日开始计算的,而生产包装上的有效期是按生产日期推算的,所以我认为生产日期应以出成品日计,批号应以成品日或流水号计.请指导一下!
答:批:为以限定数量的原材料、包装材料或应用单个工艺、或系列工艺制备的药品,具有同质性.有时需将批分为一定数量的亚批,这些亚批最后合并为具有同质性最终药品批.在终末灭菌情况下,批量由灭菌柜的容量所决定.在连续生产中,药品批必须与生产限定的工序相对应,与其同质性的要求相一致.批量既可定义为固定的数量,也可为固定时间内生产的数量.批号:在标签、批记录和相应分析证明等中,唯一鉴别批特性的清晰数字和文字的结合体.批与生产日期的联系,但不是必定联系,可用流水号进行.但生产日期要求相对严格,生产日通常是这样进行,原料药以精烘包日期为生产日期,制剂以投料日期为生产日期,生物制品通常是一连续生产过程,原料与制剂很难分别,一般以纯化后的活性物质日期为生产日期.上述是一个参考,关键对产品的稳定性试验进行考察,保证药品安全.
16 有一些作为化工原料进口的辅料,其质量优于国产的药用级辅料,工厂进厂检验也按《中国药典》进行检验,符合规定.这种情况下如果工厂使用进口辅料,是否与法规不相符.工厂应如何操作?
答:进口的辅料必须取得进口注册证后方能使用.
17 我公司准备开展GMP车间的建设,要立项吗?若要,贵局办事指南中没有相关规定.若不要,GMP初审怎么办理?
答:已经取消立项,专业设计-施工-验收-认证-投产.
18 食品原辅料的有效期是多久?哪里能够查到像油类、色素类、提取物、植物粉末类保健食品的原辅料的有效期.
答:食品标准由技术监督管,可以向他们查询.
19 我公司的许可证换证资料已上报,可现在公司营业执照企业类型由原来的“合资经营”变更为“有限责任公司”,请问药品生产许可证变更该怎么办?
答:按正常程序申请变更.
20 我想问一下:生产无菌原料药的设备比如干燥机,怎样清洁才能达到无菌的要求?
答:清洁方法很多.但直接接触药品的设备在清洁后还必须灭菌.
21 一方药品生产企业委托另一方生产一个药品,这委托方还能进行生产吗?
答:如果符合GMP条件,并取得证书,同样可以生产.
22 目前有一个膏药生产企业在河南,但为健字号,未取得药品注册及生产资格,我想在广东办一个生产企业生产此膏药产品,请问该如何办理?
答:应先办理药品注册.
23 增加厂外车间,应该在什么时候提出变更生产许可证的申请,需要准备什么资料.
答:完成新车间建设后申请,申请要求见我局办事指南中的“《药品生产许可证》变更事项”.
24 质量管理部门是否履行评价原料、中间产品及成品的质量稳定性,为确定物料贮存期、药品有效期提供数据的责任.每年每品种至少留1批进行质量稳定性评估.上面提到的“质量稳定性评估”的具体要求是什么?是必须象“长期稳定性试验”一样的作试验吗?还是可以仅仅作“加速稳定性试验”?或者是两者之外还可以设定新的稳定性评价方案?
答:与长期稳定性试验一样,对产品进行稳定性评估,包括外观、含量、相关物质等的评价,为药品有效期提供数据,是质量管理的一个重要职责.
25 关于原料的有效期,是按原料厂家生产日期起,直至制剂厂家投入生产吗?需要考虑制剂的有效期吗?另外,我们公司有些季节性的品种,最后一批剩下的原料比工艺规程所需的量多了或少了约5%或,可否一次性投进去?(在总混工序加强验证)
答:1.按现行法规,有效期算到制剂投料日期,不需要考虑其它问题.2.SOP是怎样制定,就怎样执行.
26 十万级的工作服可以降级(比如在三十万级洁净区)清洗,消毒,整理吗?
答:1.应设置不同洁净区使用的工作服清洗、整理、存放的洗衣间.2.300,000级的洁净工作服可在非洁净室(区)洗衣房内洗涤、干燥、整理.3.100,000级以上(含100,000级)区域的洁净工作服应在洁净室(区)内洗涤、干燥、整理,必要时按要求灭菌.4.10,000级以上无菌操作区使用的工作服应在10,000级洗衣间清洗、并经灭菌后送入无菌区操作.
27 我们现在由于生产规模扩大,打算在仓库区域分出一部分,按照内包装的要求,建立一个那包装车间,不知道这样是否违反GMP要求,能否实施?
答:认证完成的车间,必须按申报的平面图生产,如果对生产及产品有影响的变更,必须停产情况下进行改造,必要时需要重新认证.
28 请问,我公司正在建设厂外车间,请问应先申请变更生产许可证,还是先申请GMP认证.
答:先申请变更生产许可证,后申请GMP认证.
29 我厂是原料药生产厂家,本打算在今年12月底进行四个原料药品种的GMP认证工作,故正在准备资料先进行许可证变更.但根据厂的统一部署,认证时间可能推迟到2006年1月份,那时许可证已经换了新证,我的问题是:四个原料药品种在换证资料申报时,我厂已经作为新增生产范围上报,那么新的许可证是否含有这四个品种的生产范围.如果不含,我们明年申请GMP认证是用旧许可证申请变更还是等拿到新的许可证后再申请变更,如果拿到新许可证再申请变更,在时间上可能来不及,我们应如何操作.
答:如果是今年刚取得文号的品种,则可按新增范围品种,新证将予保留.否则按文件规定处理.
30 请问我们药厂已有胶囊剂的生产范围(已过GMP认证),能否申请缓释胶囊、避孕药胶囊的仿制?
答:缓释胶囊可以,避孕药胶囊则不行,需要独立的车间.
31 最近提到收载入"药典标准"的提取物是可以作为商品流通,如果是部颁标准或其他国家标准呢?
答:我国的标准可以,含部标.
32 多肽类冻干制剂可否在冻干粉针剂(头孢菌素类)GMP车间生产?与原产品共用一条生产线?
答:不可以.
33 某公司的许可证号为“粤hzzz———”,其生产范围指的是哪些剂型?
答:许可证编号上只是标明生产:化学药-H、中药-Z、生物制品-S、原料-y、制剂-z等的类别,不标示具体生产范围.
34 采购的中药提取物有国家标准或是地标的能否使用呢?
答:收载入药典标准的提取物是可以作为商品流通,如:冬青油等,我省南海就有一家专门生产药用油及提取物的专业厂家.
35 我公司拟生产一中药制剂,其中药提取物准备外购,供方为外省一取得药品生产许可证的企业,不知可否,需要什么手续
答:中药提取物没有标准的,不能购买,但可以委托加工,跨省的需要国家局审批.
36 退货产品重新销售是否要再全检.
答:此问题比较复杂,不是所有的退货都能销售.但重新销售,必须经检验,由授权人签字后放行.
37 我们现在只有片剂、胶囊剂的GMP证书.现在我们想开发一种冻干制剂.1、新药注册法规上面规定,需要有符合GMP条件的车间才能够拿到生产批件.如果将来我们进行委托生产,是不是这点可以不要求?2、如果我的片剂、冻干剂车间共用,每年的前半年生产片剂,后半年生产冻干剂,我们可以在做好生产车间洁净度、等等相关的管理也验证工作的条件下,得到这种不同剂型共用车间的两种剂型的GMP证书吗?了!!
答:1、向注册处咨询,2、这样不可以共用.
38 请问软膏剂和乳膏剂能不能在共用一条生产线.
答:可以.
39 生产记录用环保纸行不行(即一面已经使用过,但盖了个作废章)
答:不行.
40 请问一下,我公司是一个生产以治疗糖尿病和妇科等剂型的中成药的企业,最近引入一个治疗糖尿病的西药品种,请问可以在同一生产车间生产吗?如果可以的话,关键设备要不要分开?还有没有其它要求?
答:可以一起生产,验证证明没有互相影响,则可以共用.
41 中药的冻干粉针剂能否与化学药的冻干粉针剂在同一生产线进行?
答:可以.但特殊要求的产品则不行,如:头孢类、肿瘤类则不行.
42 在无菌原料药车间局部百级环境下生产了非无菌原料后,在经过清洗消后符合无菌原料药生产环境的情况下,第二天是否可以在同一区域下生产无菌原料药呢?
答:当然可以,生产无菌原料的车间空调系统一般是连续净化,或设置值班分机,但如果空调系统关闭,则必须重新消毒灭菌,直接接触药品的容器设备都必须灭菌后使用.
43 抗肿瘤类的生产线能否生产可用于肿瘤病人的提高免疫类的产品?
答:首先要说清楚你的产品所属类别,如果是辅助类则不可以,抗肿瘤类则可以.
44 生产、质量负责人的变更备案是否只需网上申请?还要纸质材料吗?
答:网上申请.
45 我公司最近在进行厂外车间的建设,请问是否先提出变更《药品生产许可证》,再GMP认证.
答:先提出验收申请,变更《药品生产许可证》后,再GMP认证.
46 我司是一家新开办的药品生产企业,目前厂房正在进行装修,同时新药报批工作也正在进行,我们将于十一月份取得药品生产的批准文件,按《药品管理法实施条例》我们将于取得药品生产批准文件之日起30日内提出GMP认证的申请,申请受理部门将于六个月内对我司是否符合GMP要求作出判定.我想问的是,《条例》所指的六个月内我们的产品是否可以上市销售?
答:必须同时取得药品生产许可证、GMP证书及注册文号三样东西才能销售,缺一不可.
47 我公司药品生产许可证年底到期,现正值换证期间,我公司想在生产许可证生产范围增加丸剂,请问是先进行GMP认证还是先在许可证上增加许可范围?!
答:先在许可证上增加范围,后进行GMP认证.
48 1、药品生产许可证申请注销后以什么时间生效?2、药品生产许可证申请注销后,已经生产的成品在市场上可以流通多久?流通时间限制以工商管理注销还是以税务登记注销为准?
答:以下发文件日期为准,注销后所有药品不能销售,因为企业已经不存在,已找不到执法对象.
49 企业重新认证前的硬件改造,不需要进行关键生产条件变更程序,直接验收后认证.那是不是直接递交GMP认证申请书,省局会根据具体情况派人先进行验收,然后再进行认证,还是有别的申请验收的程序呢?
答:省局会根据具体情况派人先进行验收,然后再进行认证.
50 注射用水系统(包括制备系统和分配系统)需要水循环保证水的质量.是否必须是24小时循环呢?我们实践中没有24小时循环,在每天用之前对管道蒸汽消毒,首批制的水也排放5分钟,并且经过长期的监测,水质合格,这样是否可以呢?有检查员认为我们这样不可靠,必须要24小时循环,有这样的具体规定吗?
答:必须符合下列规定:生产用注射用水应在制备后6小时内使用;制备后4小时内灭菌72小时内使用,或者在80℃以上保温、65℃以上保温循环或4℃以下存放.
51 我们新建药厂,请问一定要在拿到药品批准文号后才能申请GMP认证吗?如果提前申请行不行?如果要作进口分装是不是一定要拿到GMP证书才能申请?
答:拿到药品批准文号后才能申请GMP认证.进口分包装你可以向注册部门申请,但同样取得证书后方能生产.
52 我们是中药饮片公司,请问在哪可查到有批准文号的中药材或中药饮片呢?
答:中国药典.
53 请问硫酸阿托品、去乙酰毛花苷注射液是否属于医用毒品.
答:医疗用毒品品种范围一、毒性中药品种:(中药材和饮片)砒石(红砒、)砒霜水银生马钱子生川乌生白附子生附子生半夏生草乌生南星生巴豆斑蝥青娘虫红娘虫生甘遂生藤黄生千金子生天仙子闹羊花生娘毒红升丹雪上一只蒿白降丹蟾酥洋金花红粉轻粉雄黄二、西药毒性品种:(化学原料)去乙酰毛花甙丙阿托品洋地黄毒甙氢溴酸后马托品三氧化二砷毛果芸香碱升汞(氯化汞)水杨酸毒扁豆碱亚砷酸钾氢溴酸东莨菪碱士的年秋水仙碱硫酸长春碱硫酸醛基长春碱
54 在固体制剂药品检验中,内包装之前需进行一次化学项目的全检(我们谓之待包装检验),然后在包装的之后进行成品的检验,成品检验需做哪些项目:⑴.只要做外观包装质量检查和微生物限度两个项目,发成品检验报告时,其中的化学项目以待包装检验的结果为准;⑵.按照质量标准全检,即使前面的化学项目已经检验过,也还需要再做一次.支持第⑴种观点的人认为理论上化学项目的结果在包装之后是不会改变的,所以成品检验报告中的化学项目可以以待包装检验的结果为准;支持第⑵种观点的人认为GMP的法规要求是层层把关,所以必须全检.两种观点都有道理,哪种更合理?
答:法定上没有要求在包装前检验,此项检验可以作为产品质量控制的一个程序,但如果用作代替成品检验,则违反了统计学要求,涉及到一个随机问题,容易出现结果偏差.
55 成品检验可以不执行法定标准而按企业内控标准检验吗?比如检验报告的结论写成“本品按企业内控标准检验,结果符合规定”.当然我公司的企业内控标准比法定标准要高.
答:必须按法定标准检验,但内控标准可以作为参考,对公司产品质量进行管理.
56 请问10.1以后,GMP认证申报需要递交的资料是怎样的呢?如果是国家局的认证需要递交的资料又是怎样的呢?
答:按新的认证办法规定提交资料.
57 我厂是一间中药饮片厂,现生产一种长爪石斛属于石斛其中的一种,那么药品名称是按药典填写为石斛,还是按其它药书填写为长爪石斛呢?
答:如你们生产的品种在药典石斛项下有,则只需写石斛.
58 我们公司正在准备申报GMP认证,公司的人员象生产和质量负责人有变化,同申请拿生产许可证时不一样,公司对人员调整后需要备案吗?还是就以这次申报认证的生产和质量负责人为准呢?
答:以许可证电脑系统上备案的名单为准.
59 我公司是一位于中山国家健康基地新办制药企业,厂房建设己竣工,己向省局递交药品生产许可证的申报资料,并和一研究单位合作开发了两个中药9类品种.我公司可否在取得药品生产许可证后,将药品注册和GMP认证同时申报?如果不妥,根据我公司的实际情况,能否提供指导性方案,!
答:必须取得产品注册文号后方能认证.
60 在哪个网站或数据库或标准库能查到药品包装后密封性检查的方法及标准的信息?如泡罩包装品、铝塑袋包装品、塑料瓶+复合箔封口品等应如何检密封性?我已找过sfda、技监局、中国医药包装网等网站均无所获.
答:需要向国家标准部门技术质量监督管理局查询,他们有这样的标准.
61 我公司是一精细化工企业,现正在第一次建设某药用辅料的GMP车间,目前车间及软硬件设施还没有完成,估计在十一月下旬完成并申请验收.现请问一下GMP认证有没有规定期限
答:药用辅料还没有规定期限.
62 请问哪里可以查到报委托检验的程序和申报资料要求?
答:委托检验备案只需要提供"委托检验合同"即可,
63 药品生产企业在对进厂原辅料、包装材料的检验中,如遇使用频次较少的大型检验仪器设备(如核磁、红外等),相应的检验项目可以向具有资质的单位进行委托检验.请问具有资质的单位包括药厂吗?如果包括药厂,那对于药厂来说应具有什么资质才可接受委托检验?
答:包括药厂,在取得GMP证书后,
64 我公司办理了生产负责人变更,从省局取回两份备案表,是不是有一份要交市局备案..
答:对
65 药品生产许可证上的企业负责人,与检查条款里的“企业主管生产与质量管理的企业负责人”之间,有什么区别?如果某公司不设生产副总与质量副总,总经理下直接就是生产部部门负责人与质量部部门负责人,那么总经理会被认为是“主管生产与质量管理的企业负责人”吗?该总经理若并非相关专业人士,可否?
答:为同一概念,必须符合专业要求.
66 sfda从6月开始启用“药品生产许可证登记表填报”软件.请问现在要申请许可证许可事项或登记事项的变更,是用国家这套软件还是用原来省里自己的表格?.
答:用原来表格.
67 总平面布置图需要标明各功能间名称吗?如果车间与仓库分布在同一幢楼的不同楼层,如何在平面图上体现?另外,欲新增生产范围,申报材料上“生产、检验仪器、仪表、衡器校验情况”可以是检验计划吗?多谢!
答:各层分开标示.不可以是计划.
68 阳性对照室,可以只在万级而不设局部百级洁净操作台吗?普通不锈钢操作台可以吗?
答:我个人认为,你虽然是万级,但对于百级超净台及酒精灯下操作,这样会比前面风险要小,所以是不允许的.
69 请问处于过渡期内的新药,去年得到生产批文,能否进行非处方药转换评价工作?
答:可否转换请根据“国食药监安[2004]101号”文判断.
70 我们公司现在筹建非最终灭菌冻干粉针剂车间,但是现在有个很迷茫的问题:轧盖应该在哪个洁净级别下?有的书上说100000级,但是咱们国家局出的<药品生产验证指南>上规定的是无菌100级.应该是什么?
答:你肯定没有看法规,问题很简单,GMP附录上已经说很清楚,轧盖在100000级上进行就可以.
71 非处方药甲类品种该如何申报非处方药乙类?
答:甲乙类的区分由国家局决定,如要变更直接向国家局申请.
72 我公司是一个新开办的制药企业还没有通过GMP认证,想请教一下,我公司现在正购买新的药品回来生产,请问能否药品的申报和GMP的认证同步进行?如果能,在进行当中要注意那方面的事情.
答:不能同时进行,必须有产品才能认证.
73 我想请问原料是否每批必须全检?若在供货厂家不变的情况下可否定期全检一次?
答:不可以.
74 我公司现正在据《中国药典》2005年版修改药品包装、标签、说明书报送备案资料.听说非处方药的包装、标签、说明书中的“功能主治”仍要执行原审批备案的内容,不用按《中国药典》2005年版修改.请问是这样吗?到底那些内容要根据《中国药典》修改?那些内容仍按原备案的不变?有这方面的文件规定吗?
答:非处方药的包装、标签、说明书中的所有项目在国家局变更说明书前仍要执行原审批备案的内容,不需按《中国药典》2005年版修改.
75 我想问一下没有国家法定质量标准的中药材申请广东省药材标准备案时怎样办理?向哪个部门申请?
答:注册处.
76 我公司已通过了颗粒剂的GMP认证,现我们想增加茶剂的生产(所使用的设备与颗粒剂为共用设备),请问如何增加生产范围、是否还需再认证.
答:按办事指南程序增加即可,只对增加的范围认证.
77 中药饮片厂是否可以经营不是本厂生产的合装阿胶、合装龟甲胶等?
答:不可以.除非另领取有《药品经营许可证》.
78 我公司是一精细化工企业,现正在第一次建设某药用辅料的GMP车间,目前车间及软硬件设施还没有完成,估计在十一月下旬完成并申请验收.现请问一下GMP认证有没有规定期限?
答:没有期限,可随时申请.
79 中药饮片厂换证的生产范围是否有变更?是只填旧生产许可证的生产范围:“中药饮片”,还是按培训资料填写:正本生产范围:中药饮片(含毒性饮片),副本生产范围:中药饮片(含毒性饮片、含直接服用饮片,包括净制、切制、炒制、炙制、酒制等).
答:由当地市局核准为上报依据.
80 我公司原留样室面积太小,现计划搬迁至足够面积的留样室,是否属于关键生产条件变更?
答:此变动与生产关系不大,不用申报,给市局备案即可.
81 取得GMP证书或GMP检查之前生产的药品能否正常销售?请问GMP到期后能否生产?能否在其它不同产品生产车间生产(分别为生物和化学两个不同车间)?如果生产了能否销售?是否会被追究法律责任?
答:如果是GMP车间改选完成,验收合格后,在GMP条件下生产的药品可以在取得证书后销售.那车间证书到期,那车间必须停产.
82 我公司原外包装
设计安全有效化学产品的外部效应原则和内部效应原则的主要内容有哪些
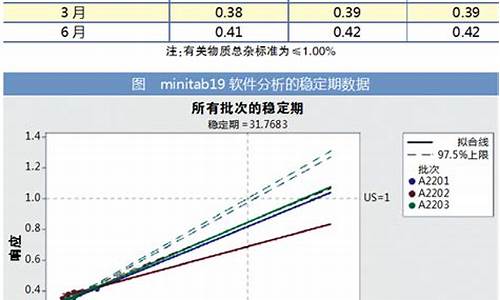
原料药的含量测定结果按百分含量表示。
原料药,指用于生产各类制剂的原料药物,是制剂中的有效成份,由化学合成、植物提取或者生物技术所制备的各种用来作为药用的粉末、结晶、浸膏等,但病人无法直接服用的物质。
化学合成药又可分为无机合成药和有机合成药。无机合成药为无机化合物(极个别为元素),如用于治疗胃及十二指肠溃疡的氢氧化铝、三硅酸镁等;有机合成药主要是由基本有机化工原料,经一系列有机化学反应而制得的药物(如阿司匹林、氯霉素、咖啡因等)。
天然化学药按其来源,也可分为生物化学药与植物化学药两大类。抗生素一般系由微生物发酵制得,属于生物化学范畴。近年出现的多种半合成抗生素,则是生物合成和化学合成相结合的产品。
原料药中,有机合成药的品种、产量及产值所占比例最大,是化学制药工业的主要支柱。原料药质量好坏决定制剂质量的好坏,因此其质量标准要求很严,世界各国对于其广泛应用的原料药都制订了严格的国家药典标准和质量控制方法。
什么是标示量,它与原料药的含量有什么区别?
化学药物质量控制分析方法验证技术指导原则
一、概述
保证药品安全、有效、质量可控是药品研发和评价应遵循的基本原则,其中,对药品进行质量控制是保证药品安全有效的基础和前提。为达到控制质量的目的,需要多角度、多层面来控制药品质量,也就是说要对药物进行多个项目测试,来全面考察药品质量。一般地,每一测试项目可选用不同的分析方法,为使测试结果准确、可靠,必须对所采用的分析方法的科学性、准确性和可行性进行验证,以充分表明分析方法符合测试项目的要求,这就是通常所说的对方法进行验证。 方法验证的目的是判断采用的分析方法是否科学、合理,是否能有效控制药品的内在质量。
从本质上讲,方法验证就是根据检测项目的要求,预先设置一定的验证内容,并通过设计合理的试验来验证所采用的分析方法能否符合检测项目的要求。
方法验证在分析方法建立过程中具有重要的作用,并成为质量研究和质量控制的组成部分。
只有经过验证的分析方法才能用于控制药品质量,因此方法验证是制订质量标准的基础。方法验证是药物研究过程中的重要内容。
二、方法验证的一般原则
原则上每个检测项目采用的分析方法,均需要进行方法验证。
方法验证的内容应根据检测项目的要求,结合所采用分析方法的特点确定。
同一分析方法用于不同的检测项目会有不同的验证要求。例如,采用高效液相色谱法用于制剂的鉴别和杂质定量试验应进行不同要求的方法验证,前者重点要求验证专属性,而后者重点要求验证专属性、准确度、定量限。
三、方法验证涉及的三个主要方面
(一)需要验证的检测项目
鉴别、
杂质检查
定量测定(含量测定、溶出度、释放度等)、
其他特定检测项目 (分子量及分子量分布、生物活性等)
鉴别的目的在于判定被分析物是目标化合物,而非其它物质,用于鉴别的分析方法要求具有较强的专属性。
杂质检查主要用于控制主成分以外的杂质,如有机杂质、无机杂质等。杂质检查可分为限度试验和定量试验两种情况。用于限度试验的分析方法验证侧重专属性和检测限。
用于定量试验的分析方法验证强调专属性、准确度和定量限。
定量测定包括含量测定、制剂的溶出度测定等,由于此类项目对准确性要求较高,故所采用的分析方法要求具有一定的专属性、准确度和线性。
其他特定检测项目包括粒径分布、旋光度、分子量分布、生物活性等,由于这些检测项目的要求与鉴别、杂质检查、定量测定等有所不同,对于这些项目的分析方法验证应有不同的要求。
(二)分析方法
分析方法是为完成上述各检测项目而设定和建立的测试方法。
分析方法原理
仪器及仪器参数
试剂
系统适用性试验
供试品溶液制备
对照品溶液制备
测定
计算及测试结果的报告等
测试方法
化学分析方法
仪器分析方法
这些方法各有特点,同一测试方法可用于不同的检测项目,但验证内容可不相
(三)验证内容
验证内容:
方法的专属性
线性
范围
准确度
精密度
检测限
定量限
耐用性
系统适用性等
四、方法验证的具体内容
(一)专属性
专属性系指在其他成分(如杂质、降解物、辅料等)可能存在下,采用的分析方法能够正确鉴定、检出被分析物质的特性。
通常,鉴别、杂质检查、含量测定方法中均应考察其专属性。如采用的方法不够专属,应采用多个方法予以补充。
1、鉴别反应
鉴别试验应确证被分析物符合其特征。
专属性试验要求证明能与可能共存的物质或结构相似化合物区分,需确证含被分析物的供试品呈正反应,而不含被测成分的阴性对照呈负反应,结构相似或组分中的有关化合物也应呈负反应。
2、杂质检查
作为纯度检查,所采用的分析方法应确保可检出被分析物中杂质的含量,如有关物质、重金属、有机溶剂等。因此杂质检查要求分析方法有一定的专属性。
在杂质可获得的情况下,可向供试品中加入一定量的杂质,证明杂质与共存物质能得到分离和检出,并具适当的准确度与精密度。
在杂质或降解产物不能获得的情况下,专属性可通过与另一种已证明合理但分离或检测原理不同、或具较强分辨能力的方法进行结果比较来确定。或将供试品用强光照射,高温,高湿,酸、碱水解及氧化的方法进行破坏(制剂应考虑辅料的影响),比较破坏前后检出的杂质个数和量。必要时可采用二极管阵列检测和质谱检测,进行色谱峰纯度检查。
3、含量测定
含量测定目的是得到供试品中被分析物的含量或效价的准确结果。 在杂质可获得的情况下,对于主成分含量测定可在供试品中加入杂质或辅料,考察测定结果是否受干扰,并与未加杂质和辅料的供试品比较测定结果。
在杂质或降解产物不能获得的情况下,可采用另一个经验证了的或药典方法进行比较,对比两种方法测定的结果。
也可采用破坏性试验(强光照射,高温,高湿,酸、碱水解及氧化),得到含有杂质或降解产物的试样,用两种方法进行含量测定,比较测定结果。
必要时进行色谱峰纯度检查,证明含量测定成分的色谱峰中不包含其他成分。
(二)线性
线性系指在设计的测定范围内,检测结果与供试品中被分析物的浓度(量)直接呈线性关系的程度。 线性是定量测定的基础,涉及定量测定的项目,如杂质定量试验和含量测定均需要验证线性。 应在设计的测定范围内测定线性关系。可用一贮备液经精密稀释,或分别精密称样,制备一系列被测物质浓度系列进行测定,至少制备5个浓度。以测得的响应信号作为被测物浓度的函数作图,观察是否呈线性,用最小二乘法进行线性回归。 必要时,响应信号可经数学转换,再进行线性回归计算,并说明依据。
(三)范围
范围系指能够达到一定的准确度、精密度和线性,测试方法适用的试样中被分析物高低限浓度或量的区间。 范围是规定值,在试验研究开始前应确定验证的范围和试验方法。
可以采用符合要求的原料药配制成不同的浓度,按照相应的测定方法进行试验。 范围通常用与分析方法的测试结果相同的单位(如百分浓度)表达。涉及到定量测定的检测项目均需要对范围进行验证,如含量测定、含量均匀度、溶出度或释放度、杂质定量试验等。
范围应根据剂型和(或)检测项目的要求确定。
1、含量测定 范围应为测试浓度的80%~100%或更宽。
2、制剂含量均匀度 范围应为测试浓度的70%~130%。根据剂型特点,如气雾剂、喷雾剂,必要时,范围可适当放宽。
3、溶出度或释放度 对于溶出度,范围应为限度的±20%;如规定限度范围,则应为下限的-20%至上限的+20%。 对于释放度,如规定限度范围为,从1小时后为20%至24小时后为90%,则验证范围应为0~110%。
4、杂质 杂质测定时,范围应根据初步实测结果,拟订出规定限度的±20%。如果含量测定与杂质检查同时测定,用面积归一化法,则线性范围应为杂质规定限度的-20%至含量限度(或上限)的+20%。
(四)准确度
准确度系指用该方法测定的结果与真实值或认可的参考值之间接近的程度。有时也称真实度。 一定的准确度为定量测定的必要条件,因此涉及到定量测定的检测项目均需要验证准确度,如含量测定、杂质定量试验等。准确度应在规定的范围内建立,对于制剂一般以回收率试验来进行验证。试验设计需考虑在规定范围内,制备3个不同浓度的试样,各测定3次,即测定9次,报告已知加入量的回收率(%)或测定结果平均值与真实值之差及其可信限。
1、含量测定
原料药可用已知纯度的对照品或符合要求的原料药进行测定,或用本法所得结果与已建立准确度的另一方法测定的结果进行比较。 制剂可用含已知量被测物的各组分混合物进行测定。如不能得到制剂的全部组分,可向制剂中加入已知量的被测物进行测定,必要时,与另一个已建立准确度的方法比较结果。
2、杂质定量试验
杂质的定量试验可向原料药或制剂中加入已知量杂质进行测定。如果不能得到杂质,可用本法测定结果与另一成熟的方法进行比较,如药典方法或经过验证的方法。 如不能测得杂质的相对响应因子,可在线测定杂质的相关数据,如采用二极管阵列检测器测定紫外光谱,当杂质的光谱与主成分的光谱相似,则可采用原料药的响应因子近似计算杂质含量(自身对照法)。并应明确单个杂质和杂质总量相当于主成分的重量比(%)或面积比(%)。
(五)精密度
精密度系指在规定的测试条件下,同一均质供试品,经多次取样进行一系列检测所得结果之间的接近程度(离散程度)。
精密度一般用偏差、标准偏差或相对标准偏差表示。取样测定次数应至少6次。
精密度可以从三个层次考察:
重复性
中间精密度
重现性
1、重复性
重复性系指在同样的操作条件下,在较短时间间隔内,由同一分析人员测定所得结果的精密度。 重复性测定可在规定范围内,至少用9次测定结果进行评价,如制备3个不同浓度的试样,各测定3次,或100%的浓度水平,用至少测定6次的结果进行评价。
2、中间精密度
中间精密度系指在同一实验室,由于实验室内部条件改变,如时间、分析人员、仪器设备、测定结果的精密度。 验证设计方案中的变动因素一般为日期、分析人员、设备。
3、重现性
指不同实验室之间不同分析人员测定结果的精密度。 当分析方法将被法定标准采用时,应进行重现性试验。
(六)检测限
检测限系指试样中的被分析物能够被检测到的最低量,但不一定要准确定量。 该验证指标的意义在于考察方法是否具备灵敏的检测能力。因此对杂质限度试验,需证明方法具有足够低的检测限,以保证检出需控制的杂质。
六、对方法验证的评价
(一)有关方法验证评价的一般考虑
总体上,方法验证应围绕验证目的和一般原则来进行,方法验证内容的选择和试验设计方案应系统、合理,验证过程应规范严谨。 并非每个检测项目的分析方法都需进行所有内容的验证,但同时也要注意验证内容应充分,足以证明采用的分析方法的合理性。如杂质限度试验一般需要验证专属性和检测限,而对于精密度、线性、定量限等涉及定量测定的项目,则一般不需要进行验证。
(二)方法验证的整体性和系统性
方法验证内容之间相互关联,是一个整体。因此不论从研发角度还是评价角度,方法验证均注重整体性和系统性。 例如,对于鉴别项目所需要的专属性,一般一种分析方法不太可能完全鉴别被分析物,此时采用两种或两种以上分析方法可加强鉴别项目的整体专属性。在方法验证内容之间也存在较多的关联性,可以相互补充。如原料药含量测定采用容量分析法时,由于方法本身原因,专属性略差,但假如在杂质检测时采用了专属性较强的色谱法,则一般认为整个检测方法也具有较强的专属性。
总之,由于实际情况较复杂,在方法验证过程中,不提倡教条地去进行方法验证。
此外,越来越多的新方法不断被用于质量控制中,对于这些方法如何进行验证需要具体情况具体分析,而不能照搬指导原则。
什么 是标示量
标示量就是制剂理论规定含主药的量,比如说某片剂,标示量为60mg,片重0.3g,就是说每片中理论上含主药60mg,它是理论值,与实际测得值有差异;原料药含量是其中所含主药的百分含量;两者的主体不同!一个是制剂,一个是原料药!
标示量,指该剂型单位剂量的制剂中规定的主药含量,简单易懂地说即是规格。分析计算时,如不将标示的规格计算进行,得到的是主药的含量;如果在分母上乘上该药的片(袋,瓶)重,分子上乘上该药的规格标示量,即得该药的标示含量。(主注意单位的换算)。
须用UV法时,可采用不受仪器及其它变化影响的“对照品比较法”定量,测定溶液的吸收度宜在0.3~0.7间。元素测定法如定氮法,在其它方法均不适宜时可采用,但因不能反映化合物含量的变化情况,此法不可用于稳定性考察中。
容量分析法的验证:
①精密度:用原料药精制品考察方法精密度,平行试验5个样本的RSD≤0.2%;
②准确度:以测定原料精制品(含量>99.5%)的回收率(测定值与理论值的比值)计算,应在99.7%~100.3%之间(n=5,RSD≤0.1%);
③滴定终点确定的依据:包括滴定曲线的绘制,如用指示剂法确定终点,应用电位法校准终点颜色,提供指示剂颜色与电位变化情况的对比结果;
④耐用性:考察测定条件(供试液稳定性、样品提取次数、时间等)有微小变动时,测定结果不受影响的承受程度,如测试条件要求苛刻时则应在方法中注明。
以上内容参考:百度百科-药品含量
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。