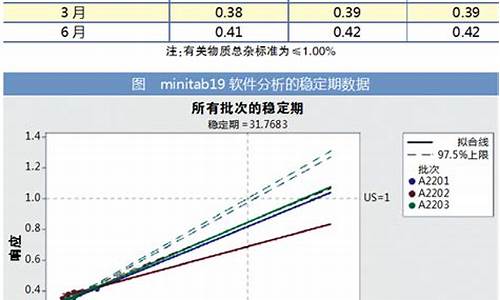
原料药注册申报的批量-原料药注册与申报

药品注册核查结果判定原则中属于不通过的情形,包括如下:
原始病历记录不详细、不完整;方案偏离未报告;个别量表的填写和修改不规范;试验用药的记录不准确安全性信息记录不完整;合并用药记录不全等。部分数据存在无法溯源、记录不完整等数据可靠性问题部分原始记录与申报资料不一致;技术转移不充分;确认与验证不充分不具备商业化生产条件等。
药品监督检查:
药品监督检查是对药品上市许可持有人、药品生产企业执行有关法律、法规,实施药品生产质量管理规范等方面开展的检查工作,核查中心按照国家药监局的工作部署,开展基于风险的监督检查。2021年,核查中心完成监督检查任务101个,包括中药监督检查任务10个、化学药品监督检查任务5个。
疫苗巡查、血液制品抽查及其他生物制品检查任务84个、麻醉精神类药品安全管理检查任务2个,对促进药品上市许可持有人和生产企业持续合规发挥了积极作用。2021年,药品监督检查不通过的任务共3个。
药品监督检查发现的主要问题包括:
质量受权人未按程序要求履行物料和产品放行职责;实际生产批量超出工艺验证批量范围;企业无法提供检验结果偏差的调查记录;企业厂房维护、设备清洁及生产过程粉尘控制措施不足,存在污染和交叉污染的风险等。
境外检查:
境外检查是对生产地在境外的药品生产企业开展的现场检查。新冠肺炎疫情发生后,由于出入境的限制,核查中心努力探索境外远程非现场的检查模式。2021年,核查中心采取境外远程非现场方式完成检查任务6个,有力保障了进口药品质量安全。2021年,境外远程非现场检查不通过的任务1个。
药品境外检查发现的主要问题包括:
在药品生产用原料、辅料和成品的质量控制方面存在严重不足;对反复、多次出现可见异物投诉情况采取的纠正与预防措施不足;未对变更进行有效控制;未对关键的生产工艺和操作规程定期进行再验证;质量协议中关于原料药放行职责约定不明确等。
原料药数据分析系统怎么样?
四稀物,小批量的可以,但必须到税务部门去备案说明,如说明是你们的副产品或废旧物资的处理等等。否则工商会查你超范围经营的问题等等。
你问的题目和内容不一致。假如是甲醇,乙醇,因为是危化品,国家规定是不容许的。你必须申领该类产品的危化品安全生产经营许可证后才可以。如果你被查到,你将面临高额的处罚。
稳定性试验指导原则
原料药对于医药产业链是上游的存在的属性,是保障药品供应、满足人民用药需求的基础,目前各国对于原料药产业的重视程度明显加大。
但是在原料药研发和使用的过程中,很多人都是“开头难、结尾也难”,为了检索药物合成路线,需要查询大量的刊期、论文、专利资料等等,耗时耗力,找到资料搞清一篇文献的合成细节,却找寻不到更多更优质的合成路线;想要购买却找寻不到合适的厂商,想要分析原料药进出口数据,分析市场动态,却没有数据来源。
对此,药融云重磅推出了原料药数据库,集成原料药生产厂商、原料药用量、海关进出口、合成路线,原料药注册信息、登记信息等,实现原料药综合查询的系统,提供全球API供需、市场规模、合成信息一站检索,帮助业内人士快速、高效的完成所需信息的查找与整理。
原料药行业数据分析系统适用于:原料药研发人员、调研人员、高校师生、医药中间体生产企业、有机化学销售员等。
一、查询合成路线,筛选最佳方案
任何一个化合物的合成路线通常都有多条,药物合成也是这样,原料药的合成,最终的目的是要走进工厂批量化的生产,选择一条适合自己生产情况的路线是至关重要。
原料药系统
药融云数据库整合了多条合成方法,包含对应的合成路线、反应条件、中间体、试剂信息。并提供了对应的参考文献,专利溯源。
1、查询合成路线
列如:在合成路线数据库中通过名称搜索“Ezetimibe”合成路线共一百多条。
药物合成路线
2、查询中间体和试剂
除了查询合成路线还可查看每条合成路线中涉及的中间体和试剂,还提供更为详细的原始资料,也可根据下方的参考文献,自行查看。
中间体试剂
二、调研市场需求、为立项/市场分析做参考
1、原料药用量推算
根据每年全球市场制剂用量推算,收载得到6000+原料药用量信息。不同品种、不同企业、不同国家市场下,各个年份中该原料药的详细用量、占比及排行;并包含全局分析模块,清晰展示各品种、区域中该产品的用量对比情况,便于探究某原料药在目标市场近年的大致用量,及分析潜在的客户、竞争企业。
调研市场需求,为立项/市场分做参考
2、查询海关进出口信息
包含了中国、美国、俄罗斯、印度等各国海关进出口货物的货物名称、时间、进口商、出口商、数量及价格等信息,可通过货物名称、海关编码、提单号检索,并支持进出口国家筛选,帮助您方便快捷地了解药物的进出口情况,进行全面市场调研、分析市场动态。
原料药海关进出口
三、生产厂商分布一键查,掌握企业供应竞争布局
在药融云原料药数据库中,可以查询目标产品的全球生产厂商分布,在中国、欧美日韩等五个国家地区的登记注册状态;掌握供应企业、竞争企业布局。
如检索「Ritonavir」,可看到该原料药在全球范围内有近50家企业可生产。中国、印度、意大利、美国等10个国家均有相应的生产企业,其中印度最多,有21家企业可生产;其次为中国,中国安徽、上海、石家庄等省市有十余家企业可生产。
掌握企业供应竞争布局
四、查询注册登记信息,满足更多需求
包含CDE原料药、药用辅料和药材包登记信息、美国DMF注册、欧盟CEP认证、日本MF注册、韩国DMF注册、FDA批准辅料等。
注册登记信息、分析更多需求
药融云原料药行业数据分析系统助力于,原料药市场分析、用量推算,查询合成路线、中间体、试剂等等,助力于业内人士快速查询相关数据。
稳定性试验的目的是考察原料药或药物制剂在温度、湿度、光线的影响下随时间变化的规律,为药品的生产、包装、贮存、运输条件提供科学依据,同时通过试验建立药品的有效期。
稳定性试验的基本要求是:
(1)稳定性试验包括影响因素试验、加速试验与长期试验。影响因素试验用一批原料药进行。加速试验与长期试验要求用三批供试品进行。
(2)原料药供试品应是一定规模生产的,供试品量相当于制剂稳定性试验所要求的批量,原料合成工艺路线、方法、步骤应与大生产一致。药物制剂供试品应是放大试验的产品,其处方与工艺应与大生产一致。药物制剂如片剂、胶囊剂,每批放大试验的规模,片剂至少应为10000片,胶囊剂至少应为10000粒。大体积包装的制剂(如静脉输液等)每批放大规模的数量至少应为各项试验所需总量的10倍。特殊品种、特殊剂型所需数量,根据情况另定。
(3)供试品的质量标准应与临床前研究及临床试验和规模生产所使用的供试品质量标准一致。
(4)加速试验与长期试验所用供试品的包装应与上市产品一致。
(5)研究药物稳定性,要采用专属性强、准确、精密、灵敏的药物分析方法与有关物质(含降解产物及其他变化所生成的产物)的检查方法,并对方法进行验证,以保证药物稳定性结果的可靠性。在稳定性试验中,应重视降解产物的检查。
(6)由于放大试验比规模生产的数量要小,故申报者应承诺在获得批准后,从放大试验转入大规模生产时,对最初通过生产验证的三批大规模生产的产品仍需进行加速试验与长期稳定性试验。本指导原则分两部分,第一部分为原料药,第二部分为药物制剂。
1.原料药
原料药要进行以下试验。
(1)影响因素试验此项试验是在比加速试验更激烈的条件下进行。其目的是探讨药物的固有稳定性、了解影响其稳定性的因素及可能的降解途径与降解产物,为制剂生产工艺、包装、贮存条件和建立降解产物分析方法提供科学依据。供试品可以用一批原料药进行,将供试品置适宜的开口容器中(如称量瓶或培养皿),摊成≤5mm厚的薄层,疏松原料药摊成≤10mm厚薄层,进行以下试验。当试验结果发现降解产物有明显的变化,应考虑其潜在的危害性,必要时应对降解产物进行定性或定量分析。
①高温试验供试品开口置适宜的洁净容器中,60℃温度下放置10天,于第5天和第10天取样,按稳定性重点考察项目进行检测。若供试品有明显变化(如含量低于规定限度)则在40℃条件下同法进行试验。若60℃无明显变化,不再进行40℃试验。
②高湿度试验供试品开口置恒湿密闭容器中,在25℃分别于相对湿度90%±5%条件下放置10天,于第5天和第10天取样,按稳定性重点考察项目要求检测,同时准确称量试验前后供试品的重量,以考察供试品的吸湿潮解性能。若吸湿增重5%以上,则在相对湿度75%±5%条件下,同法进行试验;若吸湿增重5%以下,其他考察项目符合要求,则不再进行此项试验。恒湿条件可在密闭容器如干燥器下部放置饱和盐溶液,根据不同相对湿度的要求,可以选择NaCl饱和溶液(相对湿度75%±1%,15.5~60℃),KNO3饱和溶液(相对湿度92.5%,25℃)。
③强光照射试验供试品开口放在装有日光灯的光照箱或其他适宜的光照装置内,于照度为4500lx±500lx的条件下放置10天,于第5天和第10天取样,按稳定性重点考察项目进行检测,特别要注意供试品的外观变化。
关于光照装置,建议采用定型设备“可调光照箱”,也可用光橱,在箱中安装日光灯数支使达到规定照度。箱中供试品台高度可以调节,箱上方安装抽风机以排除可能产生的热量,箱上配有照度计,可随时监测箱内照度,光照箱应不受自然光的干扰,并保持照度恒定,同时防止尘埃进入光照箱内。
此外,根据药物的性质必要时可设计试验,探讨pH值与氧及其他条件对药物稳定性的影响,并研究分解产物的分析方法。创新药物应对分解产物的性质进行必要的分析。
(2)加速实验此项试验是在规定的条件下进行。其目的是通过加速药物的化学或物理变化,探讨药物的稳定性,为制剂设计、包装、运输、贮存提供必要的资料。供试品要求三批,按市售包装,在温度40℃±2℃,相对湿度75%±5%的条件下放置6个月。所用设备应能控制温度±2℃,相对湿度±5%,并能对真实温度与湿度进行监测。在试验期间第1个月、2个月、3个月、6个月末分别取样一次,按稳定性重点考察项目检测。在上述条件下,如6个月内供试品经检测不符合制订的质量标准,则应在中间条件下即在温度30℃±2℃、相对湿度65%±5%的情况下(可用Na2CrO4饱和溶液,30℃,相对湿度64.8%)进行加速试验,时间仍为6个月。加速试验,建议采用隔水式电热恒温培养箱(20~60℃)。箱内放置具有一定相对湿度饱和盐溶液的干燥器,设备应能控制所需温度,且设备内各部分温度应该均匀,并适合长期使用。也可采用恒湿恒温箱或其他适宜设备。
对温度特别敏感的药物,预计只能在冰箱中(4~8℃)保存,此种药物的加速试验,可在温度25℃±2℃、相对湿度60%±10%的条件下进行,时间为6个月。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。