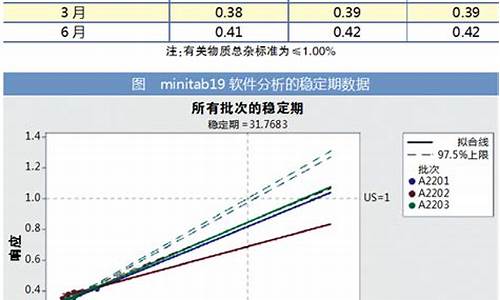
原料药含量测定首选方法那本书上规定的依据-原料药的含量测定方法的选择依据是
始自1930年出版的《中华药典》。1949年中华人民共和国成立后,已编订了《中华人民共和国药典》(简称《中国药典》)1953、1963、1977、1985、1990、1995、2000、2005、2010年版共九个版次。
1949 年10 月1 日中华人民共和国成立后,党和政府十分关怀人民的医药卫生保健工作,当年11 月卫生部召集在京有关医药专家研讨编纂药典问题。1950 年1 月卫生部从上海调药学专家孟目的教授负责组建中国药典编纂委员会和处理日常工作的干事会,筹划编制新中国药典。
1950 年4 月在上海召开药典工作座谈会,讨论药典的收载品种原则和建议收载的品种,并根据卫生部指示,提出新中国药典要结合国情,编出一部具有民族化、科学化、大众化的药典。随后,卫生部聘请药典委员49 人,分设名词、化学药、制剂、植物药、生物制品、动物药、药理、剂量8 个小组,另聘请通讯委员35 人,成立了第一届中国药典编纂委员会。卫生部部长李德全任主任委员。
1951 年4 月24 日至28 日在北京召开第一届中国药典编纂委员会第一次全体会议,会议对药典的名称、收载品种、专用名词、度量衡问题以及格式排列等作出决定。干事会根据全会讨论的意见,对药典草案进行修订,草案于1952 年底报卫生部核转政务院文教委员会批准后,第一部《中国药典》 1953 年版由卫生部编印发行。
1953 年版药典共收载药品531 种,其中化学药215 种,植物药与油脂类65 种,动物药13 种,抗生素2 种,生物制品25 种,各类制剂211 种。药典出版后,于1957 年出版《中国药典》 1953 年版第一增补本。1955 年卫生部成立第二届药典委员会,聘请委员49 人,通讯委员68 人,但这届委员会因故未能进行工作。1957 年成立第三届药典委员会,聘请委员80 人,药学专家汤腾汉教授为这届委员会主任委员(不设通讯委员),同年7 月28 日至8 月5日在北京召开第一次全体委员会议,卫生部李德全部长作了药典工作报告,特别指出第一版中国药典没有收载广大人民习用的中药,是个很大的缺陷。会议在总结工作的基础上,通过了制订药典的原则,讨论了药典的性质和作用,并修改了委员会章程,会议一致认为应把合乎条件的中药收载到药典中。8 月27日卫生部批准委员会分设药理与医学、化学药品、药剂、生化药品、生药、生物制品六个专门委员会及名词小组,药典委员会设常务委员会,日常工作机构改称秘书室。1958 年经常务委员会研究并经卫生部批准,增聘中医专家8 人、中药专家3 人组成中医药专门委员会,组织有关省市的中医药专家,根据传统中医药的理论和经验,起草中药材和中药成方(即中成药)的标准。
1959 年6 月25 日至7 月5日在北京召开这届委员会第二次全体会议,会议主要审议新版药典草稿,并确定收载品种。草稿经修订补充后,分别由各专门委员会审定,于1962 年完成送审稿,报请批准后付印,1965 年1 月26 日卫生部公布《中国药典》 1963 年版,并发出通知和施行办法。
1963 年版药典共收载药品1310种,分一、二两部,各有凡例和有关的附录。一部收载中医常用的中药材446 种和中药成方制剂197 种;二部收载化学药品667 种。此外,一部记载药品的“功能与主治”,二部增加了药品的“作用与用途”。
1966 年由于“”影响,药典委员会工作陷于停顿。1972 年4 月28 日批复卫生部“同意恢复药典委员会,四部(卫生部、燃料化学工业部、商业部、解放军总后卫生部)参加,卫生部牵头”。据此,同年5 月31 日至6 月10日在北京召开了编制国家新药典工作会议,出席会议的有全国各省(自治区、直辖市)的药品检验、药政管理以及有关.单位代表共88 人。这次会议着重讨论了编制药典的指导思想、方法、任务和要求,交流了工作经验,确定了编制新药典的方案,并分工落实起草任务。1973 年4 月,在北京召开第二次全国药典工作会议,讨论制订药典的一些原则要求,以及中西药品的标准样稿和起草说明书,并根据药材主产地和药品生产情况,调整了起草任务。1979 年10 月4 日卫生部颁布《中国药典》 1977 年版自1980 年1 月1 日起执行。1977 年版药典共收载药品1925 种。一部收载中草药材(包括少数民族药材)、中草药提取物、植物油脂以及一些单味药材制剂等882 种,成方制剂(包括少数民族药成方)270 种,共1 152 种;二部收载化学药品、生物制品等773 种。
1979 年,由卫生部聘请委员112 人组建第四届药典委员会,卫生部部长钱信忠兼任主任委员。同年11月22 日至28 日在北京召开这届第一次全体委员会议,会议讨论修改了委员会章程、药品标准工作管理办法及工作计划。委员会分设:中医、中药、医学与药理、化学药、生化药、药剂、抗生素、生物制品、放射品及名词10 个专业组。由有关专业组分别推荐新药典收载的品种,中医专业组负责审查拟定一部收载的品种范围;医学与药理专业组负责审查拟定二部收载的品种范围;由主产地所在的省(自治区、直辖市)药品检验所和有关单位负责起草标准,药典委员会办公室组织交叉复核,有些项目组成专题协作组通过实验研究后起草,标准草案经有关专业组委员并邀请有关药品检验所和药厂的代表讨论审议后报卫生部审批。《中国药典》 1985 年版于1985 年9 月出版。1986 年4 月1 日起执行。该版药典共收载药品1489 种。一部收载中药材、植物油脂及单味制剂506 种,中药成方207 种,共713 种;二部收载化学药品、生物制品等776 种。
1985 年7 月1 日《中华人民共和国药品管理法》正式执行,该法规定“药品必须符合国家药品标准或者省、自治区、直辖市药品标准”。明确“卫生行政部门颁布的《中华人民共和国药典》和药品标准为国家药品标准”。“卫生行政部门的药典委员会,负责组织国家药品标准的制定和修订”。进一步确定了药品标准的法定性质和药典委员会的任务。
1986 年卫生部根据药典委员会章程聘请委员150 人组建第五届药典委员会,由卫生部崔月犁部长兼任主任委员,常设办事机构改为秘书长制。同年5 月5 日至8 日召开第五届第一次全体委员会议,讨论修订了委员会章程,通过了“七五”期间标准工作设想,确定编制《中国药典》 1990 年版的指导思想和原则要求。分别举行中药材、中药成方制剂、化学药、抗生素、生化药及药理等专业会议,安排起草和科研任务。1987 年11 月出版《中国药典》 1985 年版增补本,新增品种23 种,修订品种172 种,附录21 项。1988 年10 月,第一部英文版《中国药典》 1985 年版正式出版。同年还出版了药典二部注释选编。1989 年3 月,各地起草的1990 年版药典标准初稿基本完成,药典委员会常设机构开始组织审稿和编辑加工。同年12 月在北京举行药典委员会主任委员、副主任委员和各专业组长扩大会议进行审议,报卫生部批准后付印。1990 年12 月3 日卫生部颁布《中国药典》 1990 年版自1991 年7 月1日起执行。
这版药典分一、二两部,共收载品种1751 种。一部收载784 种,其中中药材、植物油脂等509 种,中药成方及单味制剂275 种;二部收载化学药品、生物制品等967 种。与1985 年版药典收载品种相比,一部新增80 种,二部新增213 种(含1985 年版药典一部移人5 种);删去25 种(一部3 种,二部22 种);对药品名称,根据实际情况作了适当修订。药典二部品种项下规定的“作用与用途”和“用法与用量”,分别改为“类别”和“剂量”,另组织编著《临床用药须知》一书,以指导临床用药。有关品种的红外光吸收图谱,收入《药品红外光谱集》另行出版,该版药典附录内不再刊印。
1991 年组建第六届药典委员会,由卫生部聘请委员共168 人,卫生部陈敏章部长兼任主任委员。同年5 月16 日至18 日召开第一次全体委员会议,讨论通过了委员会的章程和编制《中国药典》 1995 年版设计方案,并成立由主任委员、副主任委员和专家共11 人组成的常务委员会。分设13 个专业组,即:中医专业组、中药材专业组、中成药专业组、西医专业组、药理专业组、化学药专业一组、化学药专业二组、化学药专业三组、抗生素专业组、生化药品专业组、生物制品专业组、放射品专业组、药品名词专业组。会后,各专业组分别召开专业组委员扩大会议,安排落实全会提出的任务。
1993 年《中国药典》 1995 年版、附录初稿发往各地作为起草、修订正文标准的依据。1994 年7 月各地基本完成了标准的起草任务,由药典委员会各专业委员会分别组织审稿工作。1994 年11 月29 日提交常务委员会扩大会议讨论审议,获得原则通过,报请卫生部审批付印。卫生部批准颁布《中国药典》 1995 年版自1996 年4 月l 日起执行。
这版药典收载品种共计2375 种。一部收载920 种,其中中药材、植物油脂等522 种,中药成方及单味制剂398 种;二部收载1 455 种,包括化学药、抗生素、生化药、放射品、生物制品及辅料等。一部新增品种142 种,二部新增品种4 ”种。二部药品外文名称改用英文名,取消拉丁名;中文名称只收载药品法定通用名称,不再列副名。编制出版《药品红外光谱集》第一卷(1995 年版)。《临床用药须知》一书经修订,随《中国药典》 1995 年版同时出版,经卫生部批准,其中的“适应证”和“剂量”部分作为药政和生产部门宣传使用和管理药品的依据。
这届药典委员会除完成1995 年版药典的编制外,还于1992 年、1993 年先后编制出版《中国药典》1990 年版第一、第二增补本,二部注释和一部注释选编,《中药彩色图集》和《中药薄层色谱彩色图集》以及《中国药品通用名称》等标准方面的配套丛书。《中国药典》 1990 年版英文版亦于1993 年7 月出版发行。
为加强国家药品标准工作,1993 年5 月21 日卫生部决定将药典委员会常设机构从中国药品生物制品检定所分离出来,作为卫生部的直属单位,这是药典委员会机构史上一次重大的改革。
1996 年5 月经卫生部批准,第七届药典委员会成立,由卫生部聘请204 位委员组成,其中名誉委员18 人,卫生部陈敏章部长兼任主任委员.1998 年9 月,根据中编办(1998 ) 32 号文:卫生部药典委员会更名为国家药典委员会,并成建制划转国家药品监督管理局管理。因管理体制的变化及1999 年3 月陈敏章部长逝世,在征得有关领导部门同意后,按照第七届药典委员会章程精神,经1999 年12 月第七届药典委员会常务委员会议一致同意调整主任委员、副主任委员。这届委员会设专业委员会共16 个,分别为:中医专业委员会、中药第一专业委员会、中药第二专业委员会、中药第三专业委员会、中药第四专业委员会、医学专业委员会、药品名词专业委员会、附录专业委员会、制剂专业委员会、药理专业委员会、化学药品第一专业委员会、化学药品第二专业委员会、抗生素专业委员会、生化药品专业委员会、放射品专业委员会、生物制品专业委员会。
1996 年召开第七届药典委员会常务委员会第一次会议,通过了这届药典委员会提出的“《中国药典》 2000年版设计方案”,一部确立了“突出特色,立足提高”,二部确立了“赶超与国情相结合,先进与特色相结合”的指导思想。根据这届委员会提出的设计方案,1996 年10 月起,各专业委员会先后召开会议,落实设计方案提出的任务并分工进行工作。1997 年底,首先完成了附录与制剂通则的修改,并下发各起草单位征求意见。1998 年底药典初稿完成,经进一步征求全国各有关方面的意见,至1999 年10 月底,先后召开了16 个专业委员会审定稿会议。《中国药典》 2000 年版于1 999 年12 月经第七届药典委员会常务委员会议审议通过,报请国家药品监督管理局批准颁布,于2000 年1月出版发行,2000 年7 月1 日起正式执行。
2000 年版药典共收载药品2691 种,其中一部收载992 种,二部收载1699 种。一、二两部共新增品种399 种,修订品种562 种。这版药典的附录作了较大幅度的改进和提高,一部新增附录10 个,修订附录31 个;二部新增附录27 个,修订附录32 个。二部附录中首次收载了药品标准分析方法验证要求等六项指导原则,对统一、规范药品标准试验方法起指导作用。现代分析技术在这版药典中得到进一步扩大应用。第七届药典委员会还完成了《中国药典》 1995 年版一九九七年增补本、一九九八年增补本、《中国药品通用名称》(一九九八年增补本)及《药品红外光谱集》(第二卷)、《临床用药须知》(第三版)。1997 年完成了《中国药典》 1995 年版英文版。为加强国际合作与交流,第七届药典委员会还决定《中国药典》 2000 年版英文版与中文版同步出版。
以往几版药典中的“剂量”、“注意”项内容,由于过于简单不能准确反映临床用药的实际情况,根据“《中国药典》 2000 年版设计方案”的提议,这版药典二部取消了这两项,其有关内容移至《中国药典》 2000 年版《临床用药须知》一书中。
2002 年10 月经国家药品监督管理局(2003 年9 月更名为国家食品药品监督管理局)批准,第八届药典委员会成立。由国家药品监督管理局聘请312 位委员组成,不再设立名誉委员。国家药品监督管理局局长郑筱英兼任主任委员。原常务委员会更名为执行委员会,由全体委员大会授权审定《中国药典》及国家药品标准的重大事项。本届委员会设专业委员会共24 个。在上一届委员会的基础上,增设了民族药专业委员会(筹)、微生物专业委员会、药品包装材料与辅料专业委员会;原生物制品专业委员会扩增为血液制品专业委员会、病毒制品专业委员会、细菌制品专业委员会、体细胞治疗与基因治疗专业委员会、重组制品专业委员会和体外诊断用生物试剂专业委员会。
2002 年10 月召开第八届药典委员会全体大会及执行委员会第一次会议,通过了本届药典委员会提出的“《中国药典》 2005 年版设计方案”。设计方案明确了坚持继承与发展、理论与实际相结合的方针;确定了“科学、实用、规范”等药典编纂原则;决定将《中国生物制品规程》并人药典,设为药典三部;并编制首部中成药《临床用药须知》。
2002 年11 月起,各专业委员会先后召开会议,安排设计方案提出的任务并分别进行工作。2003 年7月,首先完成了附录草案,并发有关单位征求意见。2004 年初药典附录与品种初稿基本完成,增修订内容陆续在国家药典委员会网站上公示3 个月,征求全国各有关方面的意见。6 月至8 月,各专业委员会相继召开了审定稿会议。9 月,《中国药典》 2005 年版经过第八届药典委员会执行委员会议审议通过,12 月报请国家食品药品监督管理局批准颁布,于2005 年1 月出版发行,2005 年7 月1 日起正式执行。本版药典收载的品种有较大幅度的增加。共收载3214 种,其中新增525 种。药典一部收载品种1146 种,其中新增154 种、修订453 种;药典二部收载1967 种,其中新增327 种、修订522 种;药典三部收载101 种,其中新增44 种、修订57 种。《中国药典》 2000 年版收载而本版药典未收载的品种共有9 种。2000 年版《中国生物制品规程》及2002 年增补本收载而未收载人药典的品种共有123 种。
本版药典收载的附录,药典一部为98 个,其中新增12 个、修订48 个,删除1 个;药典二部为137 个,其中新增13 个、修订65个、删除1 个;药典三部为140 个,其中新增62 个、修订78 个,删除1 个。一、二、三部共同采用的附录分别在各部中予以收载,并进行了协调统一。
本版药典在主任委员的积极倡导下,对药品的安全性问题更加重视。药典一部采用原子吸收和电感耦合等离子体质谱法增加了有害元素(铅、镉、砷、汞、铜)测定法,并规定了有害元素的限度;药典一部还增加了中药注射剂安全性检查法应用指导原则。药典二部有126 个静脉注射剂增订了不溶性微粒检查,增修订细菌内毒素检查的品种达112 种;残留溶剂测定法中引入国际间已协调统一的有关残留溶剂的限度要求,并有24 种原料药增订了残留溶剂检查;药典二部还增加了药品杂质分析指导原则、正电子类和锝[ 99mm Tc ]放射品质量控制指导原则。药典三部增订了逆转录酶活性检查法、人血白蛋白铝残留量测定法等,牛血清白蛋白残留量及CHO 细胞蛋白残留量等检测方法也得到改进。本版药典结合我国医药工业的现状和临床用药的实际情况,将由卫生部颁布的原《澄明度检查细则和判断标准》修订为“可见异物检查法”,以加强注射剂等药品的用药安全。
本版药典坚持注重环保的一贯性原则,在品种中对苯等有害溶剂,尽可能采用其他溶剂替代。本版药典根据中医辨证施治的理论,对收载的中成药标准项下的功能与主治进行了科学规范,为准确理解中成药的功能主治及合理用药提供了保证,促进中医药在新时期的健康发展。
本版药典三部源于《中国生物制品规程》。自1951 年以来,该规程已有六版颁布执行,分别为1951 年及1952 年修订版、1959 年版、1979 年版、1990 年版及1993 年版(诊断制品类)、1995 年版、2000 年版及2002 年增补本。2002 年翻译出版了第一部英文版《中国生物制品规程》(2000 年版)。
第八届药典委员会还完成了《中国药典》 2000 年版2002 年增补本、2004 年增补本、《中国药品通用名称》(2005 年版)及《药品红外光谱集》(第三卷)、《临床用药须知》(中成药第一版、化学药第四版)。2005 年,完成了《中国药典》 2005 年版英文版.为加强国际合作与交流,本届委员会期间,与美国药典委员会联合举办了首届中美药典论坛。
为加强和提高国家标准工作效率与水平,常设机构完成了办公自动化及标准数据库的建设,实现了已颁布标准的计算机网络检索查询与统计分析。
《中国药典》的特色之一即在于它继承发扬了传统医药学的成果,并实现了中西医药学的结合。
2010版《中国药典》将于2010年10月1日正式实施。
台湾于1949年和1980年也先后出版了《中华药典》第二版和第三版。《中华药典》第三版收载各类药品746种。 (一).凡例
1.名称与编排
2.检验方法与限度
3.标准品、对照品
4.精确度
(二)正文
名称(英文和汉语拼音),结构式,性状鉴别、检查、含量测定,类别,贮藏,制剂
(三)附录
组成:制剂通则、检测方法和指导原则、滴定液的配制与标定等
(四)索引
一部:中文、汉语拼音、拉丁名、拉丁学名
二部:中文索引: 英文索引:
三部:中文索引: 英文索引:
药物分析笔记:药物分析的任务与发展
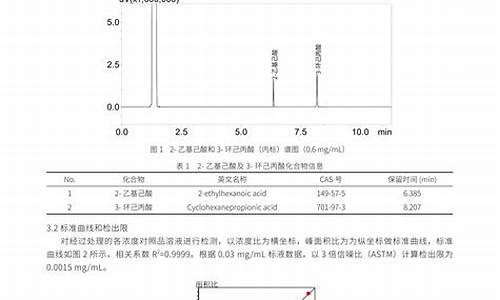
方法的验证: 订入质量标准的含量测定法不同于一般质量考察的方法,须经过严格的方法学验证,不同原理的测定法具有不同的验证内容及要求:
(1)容量分析法的验证:①精密度:用原料药精制品考察方法精密度,平行试验5个样本的RSD≤0.2%;②准确度:以测定原料精制品(含量>99.5%)的回收率(测定值与理论值的比值)计算,应在99.7%~100.3%之间(n=5,RSD≤0.1%);③滴定终点确定的依据:包括滴定曲线的绘制,如用指示剂法确定终点,应用电位法校准终点颜色,提供指示剂颜色与电位变化情况的对比结果;④耐用性:考察测定条件(供试液稳定性、样品提取次数、时间等)有微小变动时,测定结果不受影响的承受程度,如测试条件要求苛刻时则应在方法中注明。
(2)HPLC法的验证:①精密度:RSD≤2%(n=5);②准确度:用于制剂时,要考察辅料的影响,将一定量药物加到按处方比例配制的辅料中(为标示量的80%~120%)制成高、中、低三个剂量,混合均匀后,每个剂量取三份样品,按拟定方法测定回收率,应在98%~102%之间(n=9, RSD≤2%)。③线性范围:用已知含量的精制品配制一系列浓度的溶液(n=5~7),用浓度C对峰面积A或峰或被测物的响应值之比进行回归处理,线性方程的相关系数r≥0.999,截距应趋于零,并提供线性关系图;④专属性:辅料、有关物质或降解产物峰对主药峰应无干扰;⑤耐用性:考察测定条件(供试液稳定性、流动相组成和pH值、不同品牌或批号的同类色谱柱、柱温、流速、样品提取次数、时间等)有微小变动时,测定结果不受影响的承受程度,如测试条件要求苛刻时则应在方法中注明;⑥灵敏度:作为常量分析法,此项可不作主要要求。
(3)UV法的验证:①精密度:RSD≤1%(n=5);②准确度:方法同HPLC法,回收率应在98%~102%之间(n=9, RSD≤2%),同时要求辅料、有关物质或降解产物在测定波长处无吸收。③线性范围:用已知含量的精制品配制一系列浓度的溶液(n=5~7,吸收度A在0.2~0.7间),用浓度C对峰面积A或峰或被测物的响应值之比进行回归处理,线性方程的相关系数r应≥0.999,截距应趋于零,并提供线性关系图;④耐用性:考察测定条件(供试液稳定性、样品提取次数、时间、比色法中显色剂用量、反应温度、时间、pH值等)有微小变动时,测定结果不受影响的承受程度,如测试条件要求苛刻时则应在方法中注明;⑤灵敏度:作为常量分析法,此项可不作主要要求。
吸收度A在0.2~0.8间其线形关系好,利用标准品建立标准曲线后,受测溶液做适当的稀释,使其吸收度在0.2~0.8,如果太小或太大,都将影响测定的准确性的。
原料药与药物制剂的质量控制在方法的选择上有什么异同?
第一章 药物分析的任务与发展
药物分析是一门研究药品及其制剂的组成、理化性质、真伪鉴别、纯度检查及其有效成分的含量测定等的一门学科。目的是保证人们用药安全、合理、有效。
药品用于防病、治病、诊断疾病、改善体质、增强机体抵抗力的物质。
药典是一个国家关于药品标准的法典,是国家管理药品生产与质量的依据。医学教育网
第2章 药物分析的基础知识
第一节 药品检验工作的基本程序
一般为取样、鉴别、检查、含量测定、写出报告。
取样:
鉴别:判断真伪。 检查:称纯度检查,判定药物优劣。 含量测定:测定药物中有效成分的含量。
检验报告必须明确、肯定、有依据。
计量仪器认证要求:县级以上人民政府计量行政部门负责进行监督检查。符合经济合理、就地就近。
第二节 药品质量标准分析方法验证
目的是证明采用的方法适合于相应的检测要求。医学教育网
验证内容:准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐用性。
一、准确度: 是指用该方法测定的结果与真实值或参考值接近的程度,一般以百分回收率表示。
至少用9次测定结果进行评价。
二、精密度: 是指在规定的条件下,同一个均匀样品,经过多次取样测定所得结果之间的接近程度。
用偏差、标准偏差或相对标准偏差表示。
1、 重复性:相同条件下,一个分析人员测定所得结果的精密度称为重复性。至少9次。
2、 中间精密度:同一个实验室,不同时间不同分析人员用不同设备测定结果的精密度。
3、重现性:不同实验室,不同分析人员测定结果的精密度。分析方法被法定标准采用应进行重现性试验。
三、专属性:指在其他成分可能存在的情况下,采用的方法能准确测定出被测物的特性,用于复杂样品分析时相互干扰的程度。鉴别反应、杂质检查、含量测定方法,圴应考察专属性。医学,教育网
四、检测限:指试样中被测物能被检测出的最低量,无须定量。用百分数、ppm或ppb表示。
五、定量限:指样品中被测物能被定量测定的最低量,测定结果应具一定的精密度和准确度。
六、线性:系指在设计的范围内,测试结果与试样中被测物浓度直接呈正比关系的程度。
七、范围:能达到一定的精密度、准确度和线性的条件下,测试方法适用的高低限浓度或量的区间。
八、耐用性:指在一定的测定条件稍有变动时,测定结果不受影响的承受程度。
第三节 药物分析的统计学知识医学教育.网
测量误差:测量值和真实值之差。 绝对误差和相对误差。
真实值:是有经验的人用最可靠的方法对试样进行多次测定所得的平均值。
系统误差:(1)方法误差 (2)试剂误差 (3)仪器误差 (4)操作误差
偶然误差:不可定误差或随机误差,由偶然原因引起。可增加平得测定次数。
测量值的准确度表示测量的正确性,测量值的精密度表示测量的重现性。精密度是表示准确度的先决条件,只有在消除了系统误差后,才可用精密度同时表达准确度。
提高分析准确度方法:
1、选择合适的分析方法
2、减少测量误差
3、增加平行测定次数
4、消除测量过程中的系统误差(校准仪器、做对照试验、做回收试验、做空白试验)
有效数字的处理:0.05060g是四位有效数字。首位是8或9,有效数字可多记一位。PH=8.02是两位有效数字。四舍六入五成双原则。修约标准偏差或其他表示不确定度时,修约结果可使准确度估计值变得差一点。S=2.13——2.2 G检验法、4d法,>舍去。
第四节 药品质量标准制定的原则和基本内容
原则:安全有效,技术先进,经济合理。 检验方法:准确、灵敏、简便、快速。
(一)名称:
(二)性状:医学教育网;
1、外观、臭、味和稳定性
2、溶解度:一定程度上反映药品的纯度。
3、物理常数
(1)馏程:2000规定:在标准压力(101.3kPa)下,按药典装置,自开始馏出的第五滴算起,至供试品仅剩3-4ml或一定比例的容积馏出时的温度范围。
(2)熔点:系指一种物质固体熔化成液体的温度,熔融同时分解的温度,或在熔化时自初熔至全熔的一段温度。
(3)凝点:系指一种物质由液体凝结为固体时,在短时间内停留不变的温度。
(4)比旋度:具光学异构体分子的药物,旋光性能不同。按干燥品或无水物计算。准确至0.01 .
(5)折光率:光线自一种透明介质进入另一种透明介质时,两种介质密度不同,光的进行速度发生变化,即发生折射现象,遵从折射定律。对于液体药品,尤其是植物油,检查药品的纯杂程度,测定溶液的浓度。
(6)粘度:流体对流动的阻抗能力。共三法,毛细管内径。
(7)吸收系数:物质对光的选择性吸收波长。
(三) 鉴别:用理化方法或生物学方法来证明药品真实性的方法。对已知物。
(四) 杂质检查:有效性,纯度要求和安全性。
1、有效性试验
2、酸碱度 医学教育网
3、溶液的澄清度与颜色
4、无机阴离子:氯化物和硫酸盐。
5、有机杂质
6、干燥失重和水分
7、炽灼残渣:指硫酸化灰分,用于考察有机药物中混入的无机杂质。一般限度为0.1% .
8、金属离子和重金属检查 每日剂量0.5g——以上且长期服用的品种。
9、硒和砷:硒检查有:醋酸地塞米松、醋酸曲安奈德及醋酸氟轻松。第一法:古蔡氏法
10、安全性检查
(五)含量测定或效价测定: 理化方法称含量测定 生物学方法或生化方法测定称效价测定。
1、容量分析法:化学原料药含量测定的首选法。
中和法、非水滴定法、银量法、络合法、碘量法、重氮化法。
2、重量法:精密度好准确度高, 繁琐,不能应用容量法时用。挥发法、萃取法、沉淀法
3、紫外分光光度法:简便、快速。原料药避免。
4、气相色谱法:分离效果优越,
5、对含杂质和挥发性的原料药效好。维生素E医学教育网
6、高效液相色谱法:用于多组分抗生素,生化药品或因杂质干扰测定。常规方法又难分离药品。
在药典上可以清晰的看出,由于制剂可能有辅料的干扰,所以一般尽量用高效液相来进行含量测定,而原料则可以用滴定法或紫外、荧光等来检测含量。但对于那些杂质紫外吸收和药品紫外吸收波长相同的原料也要用高效液相色谱法。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。