原料药杂质研究指导原则-原料药杂质研究

有,需要继续控制。原料药是原料药,制剂是制剂。你知道这些杂质是原料药中带入的,只能证明了它的来源。现在对于杂质的要求非常严格,即便是原料药中的杂质也分为:起始物料、中间体或者降解产物等等。看这个吧,超过了报告限度的杂质,是需要出报告的。超过鉴定限度的杂质是需要知道它具体的化学结构的。尤其是你在制剂研究、生产过程中监控这些杂质的增长情况。有些杂质在原料药里增长不明显,做成了制剂就蹭蹭地长。到底是湿度影响,还是酸碱度影响,或者是温度影响。这关系到制剂工艺的问题,另外产品贮藏条件,包装条件都是跟主成分含量以及杂质有关系的。
原料药的鉴别也是对杂质的检查,对吗
没明白你的问题
你是想问:为什么做制剂的过程中,原料药中的杂质含量会增高?
还是为什么制剂的杂质含量限度比原料药高?
反正这么说吧,我不知道你是哪一种药物。但是一般的药物都会有一些不稳定的因素。比如光照、高温、高湿、酸、碱、氧化。在制剂过程中,有些是不能避免的。
比如你湿法制粒,那么肯定原料药会遇到水,然后烘干过程肯定会经过高温。诸如此类的步骤,导致了原料药中的杂质增高。所以一般来说,制剂的杂质比原料药的含量高一些属于正常现象。制剂的杂质限度也会比原料药的杂质限度宽松一些。
一般杂质的检查方法均在<中国药典>()加以规定
这个选项不对。
鉴别主要是看这个药品是不是说明书上所标注的主成分。是针对主成分的鉴别。你写的头孢,你的药品也确实是头孢。不能duang地一下变成四环素。
一般原料药的鉴别是一个紫外(或者高效液相)鉴别,一个红外鉴别,一个化学鉴别。
这三项都是为了确保这个药品与对照品一致。而且这三个鉴别根本看不出杂质。
杂质检查在检查项的有关物质里面。
EDQM和FDA对于原料药中的元素杂质怎么控制
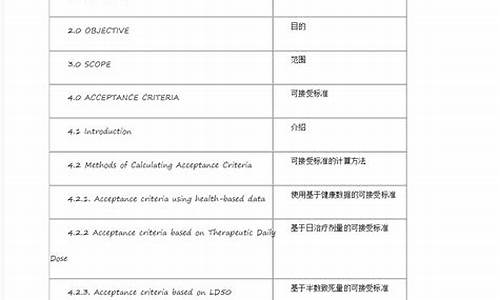
本原则用于指导化学合成的原料药及其制剂的杂质分析,并供药品研究、生产、质量标准起草和修订参考。本原则不涵盖生物/生物技术制品、肽、寡聚核苷酸、放射品、发酵产品与其半合成产品、中药和来源于动植物的粗制品。检查对象明确为某一物质时,以该杂质的化学名作为检查项目名称,如磷酸可待因中的“吗啡",氯贝丁酯中的“对氯酚”,盐酸苯海索中的“哌啶苯丙酮”,盐酸林可霉素中的“林可霉素B”和胰蛋白酶中的“糜蛋白酶"等。如果该杂质的化学名太长,又无通用的简称,可参考螺内酯项下的“巯基化合物”、肾上腺素中的“酮体”、盐酸地芬尼多中的“烯化合物”等,选用相宜的名称。在质量标准起草说明中应写明已明确杂质的结构式。
杂质是药品的关键质量属性,可影响产品的安全性和有效性。药品质量标准中的杂质系指在按照经国家药品监督管理部门依法审查批准的工艺和原辅料生产的药品中,由其生产工艺或原料带入的杂质,或在贮存过程中产生的杂质,不包括变更生产工艺或变更原辅料而产生的新杂质,也不包括掺入或污染的外来物质。若药品生产企业变更生产工艺或原辅料引入新的杂质,则需要对原质量标准进行修订,并依法向药品监督管理部门申报批准。药品中不得掺入其组分以外的物质或污染药品。对于假药和劣药,必要时应根据具体情况,采用合适的且经过验证的分析方法予以检测。
药品杂质通常分为:有机杂质、无机杂质、残留溶剂。有机杂质可在药品的生产或贮存中引入,也可由药物与辅料或包装结构的相互作用产生,这些杂质可能是已鉴定或者未鉴定的、挥发性的或非挥发性的,包括起始物、副产物、中间体、降解产物、试剂、配位体和催化剂;其中化学结构与活性成分类似或具渊源关系的有机杂质,通常称为有关物质。无机杂质可能来源于生产过程,如反应试剂、配位体、催化剂、元素杂质、无机盐和其他物质(例如:过滤介质,活性炭等),一般是已知和确定的。药品中的残留溶剂系指原料药或辅料的生产中,以及制剂制备过程中使用的,但在工艺操作过程中未能完全去除的有机溶剂,一般具有已知的毒性。
获得FDA认证的程序
对于原料药来说,通过FDA批准主要有两个阶段:一是DMF文件的登记,要求递交的DMF文件对所申请的药品的生产和质量管理的全过程以及药品质量本身做一个详尽的描述。FDA要为此文件保密,该文件是由FDA的药物评价及研究中心(Center for Drug Evaluation and Research, CDER)来审核。二是当DMF文件的登记已经完成,而且在美国的原料药品终端用户提出了申请以后,FDA官员对原料药物的生产厂家进行GMP符合性现场检查,通过对药品生产全过程的生产管理和质量管理状况的全面考察,做出该原料药生产企业的生产和质量管理能否确保所生产药品的质量的判断。FDA在现场检查的基础上做出是否批准该原料药品在美国市场上市的决定。
其基本程序如下所示:
1.进行国际市场调研,摸清美国市场目前的销售情况,对市场发展趋势与走向做出正确的预测、分析和判断,选择好申请FDA批准的品种。
2.选择申请代理人和代理经销商,并签订委托协议书、签署委托书。
3.编写申请文件,原料药为DMF文件,由代理人完成申请文件终稿的编写并向FDA递交,取得DMF文件登记号。
4.FDA收到申请文件后,经初审合格后发通知函给申请人,并发给一个登记号,说明DMF文件持有人的责任和义务。
5.工厂按美国cGMP的要求进行厂房、设施设备的改造和并完善生产质量管理的各项软件和相关人员的强化培训。
6.应美国制剂生产厂家(即该原料药品的终端用户)的申请, FDA派官员到生产厂家按照FDA颁布的生产现场检查指南并对照已上报审核的DMF文件进行检查,FDA官员在生产现场的基础上出具书面意见给生产厂家并向FDA报告检查结果。
7.FDA审核批准后将审核结果通知生产厂家并输入美国海关的管理系统,该原料药品即获准直接进入美国市场。
8.生产厂家每年向FDA递交一份DMF修改材料,一般情况下, 每2~3年可能要接受一次复查。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。