原料药登记的a和i表示什么意思-原料药级别分类
进口药品管理办法
(国家药品监督管理局令第6号)
第一章 总 则
第一条 为加强进口药品的监督管理,保证进口药品的质量和安全有效,根据《中华人民共和国药品管理法》的规定,制定本办法。
第二条 国家对进口药品实行注册审批制度。进口药品必须取得中华人民共和国国家药品监督管理局核发的《进口药品注册证》,并经国家药品监督管理局授权的口岸药品检验所检验合格。
第三条 国家药品监督管理局主管进口药品的审批和监督管理工作,地方各级药品监督管理部门主管辖区内进口药品的监督管理工作。
第四条 进口药品必须符合《中华人民共和国药品管理法》和中国其它有关法律法规的规定,必须接受国家药品监督管理局对其生产情况的监督检查。
第五条 进口药品必须是临床需要、安全有效、质量可控的品种。
第二章 申报和注册审批
第六条 申请注册的进口药品必须获得生产国国家药品主管当局注册批准和上市许可。
第七条 进口药品的生产厂必须符合所在国药品生产质量管理规范和中国药品生产质量管理规范(GMP)的要求,必要时须经国家药品监督管理局核查,达到与所生产品种相适应的生产条件和管理水平。
第八条 进口药品注册,须由国外制药厂商驻中国的办事机构或其在中国的注册代理提出申请,填写《进口药品注册证申请表》,连同本办法规定的资料,报国家药品监督管理局审批。
国外制药厂商驻中国的办事机构或其在中国的注册代理必须是在中国工商行政管理部门登记的合法机构。
第九条 申请进口药品注册,须报送以下资料:
(一)药品生产国国家药品主管当局批准药品注册、生产、销售、出口及其生产厂符合药品生产质量管理规范(GMP)的证明文件和公证文件。
(二)国外制药厂商授权中国代理商代理申报的证明文件,中国代理商的工商执照复印件;国外制药厂商常驻中国代表机构登记证复印件。
(三)药品专利证明文件。
(四)药品生产国国家药品主管当局批准的药品说明书。
(五)药品质量标准和检验方法。
(六)药品各项研究结果的综述。
(七)药品处方、生产工艺、药理、毒理及临床研究等详细技术资料。
(八)药品及包装实样和其他资料。
申报资料的具体要求,按照本办法所附《进口药品申报资料细则》(附件一)的规定执行。
第十条 申请注册的进口药品,必须按照本办法所附《进口药品质量复核规则》(附件二)的要求和程序进行质量复核。
第十一条 申请注册的进口药品,必须按照国家药品监督管理局规定的程序和要求在中国进行临床研究(包括生物等效性试验)。其中申请注册的原料药若中国尚未生产,则应用该原料药制成的制剂在中国进行临床研究。临床研究须按照中国《新药审批办法》及《药品临床试验管理规范》(GCP)的规定执行。特殊病种或其它情况需减免临床研究的,需经国家药品监督管理局审查批准。
第十二条 申报品种的质量复核和临床研究结束后,国家药品监督管理局对有关资料进行审查,符合要求的,核发《进口药品注册证》。该品种的质量标准即成为进口药品注册标准,中文说明书为指导进口药品在中国临床使用的法定说明书。
第十三条 对特殊病种的治疗药物,在中国尚没有其它替代药物的情况下,国家药品监督管理局可采取加快审批措施。
第十四条 中国重大灾情、疫情所需药品,临床特需、急需药品,捐赠药品和研究用样品等,在尚未取得《进口药品注册证》的情况下,可经国家药品监督管理局特别批准进口。此类药品仅限在特定范围内用于特定目的。
第十五条 下列情形之一的药品,其进口注册申请将不予批准:
(一)不符合本办法第六条和第七条规定的;
(二)申报资料不符合中国进口药品注册审批要求的;
(三)临床使用中存在严重不良反应的;
(四)临床疗效不确切或所报临床研究资料无法说明药品的确切疗效的;
(五)质量标准及检验方法不完善,质量指标低于中国国家药品标准、中国生物制品规程或国际通用药典以及已注册同类品种的企业标准的;
(六)含有中国禁止进口的成份的;
(七)其他不符合中国有关法律、法规和规定的。
第三章 进口药品注册证
第十六条 《进口药品注册证》是国家药品监督管理局核发的允许国外生产的药品在中国注册、进口和销售使用的批准文件。国家药品监督管理局各口岸药品检验所凭《进口药品注册证》接受报验。
第十七条 《进口药品注册证》分为正本和副本,自发证之日起,有效期三年。
第十八条 《进口药品注册证》按统一格式编号,为注册证号。注册证号由字母X(Z或S)后接八位阿拉伯数字组成,前四位为公元年号,后四位为年内顺序编号;其中Z代表中药,S代表生物制品,X代表化学药品。
第十九条 《进口药品注册证》规定以下内容:药品通用名称、商品名、主要成分、剂型。规格、包装规格、药品有效期;公司、生产厂名称及地址;注册证有效期、检验标准、注册证证号、批准时间、发证机关及印鉴等。
批准注册品种的每个不同规格,分别核发《进口药品注册证》;每个《进口药品注册证》最多登载二个包装规格。
第二十条 《进口药品注册证》只对载明的内容有效,其任何内容的改变必须报国家药品监督管理局审核批准。
第二十一条 对部分批准进口注册的原料药、辅料、制剂半成品,国家药品监督管理局将在《进口药品注册证》备注中,限定其使用范围。
第四章 《进口药品注册证》的换发和审批
第二十二条 换发《进口药品注册证》应由国外制药厂商驻中国的办事机构或其在中国的注册代理在注册证期满6个月前,向国家药品监督管理局提出申请。超过注册证有效期的按新申请注册品种管理。
第二十三条 申请换发《进口药品注册证》须填写《换发进口药品注册证申请表》,并报送以下资料:
(一)药品生产国国家药品主管当局签发的批准药品注册、生产、销售、出口及符合药品生产质量管理规范(GMP)的证明文件。
(二)国外制药厂商授权中国代理商代理申报的证明文件,中国代理商的工商执照复印件;国外制药厂商常驻中国代表机构登记证复印件。
(三)药品生产国国家药品主管当局批准的药品说明书及其中文译本。
(四)药品处方、生产工艺、质量标准及检验方法。
(五)《进口药品注册证》有效期内在中国进口、销售情况(包括进口批次、数量、进口口岸等)。
(六)进口药品使用及不良反应情况的总结报告。
(七)药品及包装实样和其他资料。
申报资料的具体要求,按《进口药品申报资料细则》的规定执行。
第二十四条 药品处方中辅料、生产工艺、质量标准和说明书等有变化的,须同时报送以下资料:
(一)修改理由及其说明。
(二)生产国国家药品主管当局批准此项修改的证明文件。
(三)此项修改所依据的实验研究资料。
第二十五条 国家药品监督管理局对申请换发《进口药品注册证》的品种进行审查,必要时,可安排质量考核或临床再评价,符合要求的,批准换发《进口药品注册证》,发给新的注册证号。
第二十六条 有下列情形之一的进口药品,其换发《进口药品注册证》申请将不予批准:
(一)发现严重不良反应的;
(二)临床疗效不确切、质量不稳定的;
(三)口岸检验二批不合格的;
(四)已被国家药品监督管理局处罚二次以上的(含二次);
(五)其他不符合中国有关法律、法规和规定的。
第五章 补充申请
第二十七条 已取得《进口药品注册证》的进口药品,下列情形属补充申请。
(一)《进口药品注册证》注明的通用名称、商品名、公司名称、生产厂名称等改变。
(二)质量标准、生产工艺、有效期等改变。
(三)适应症增加。
(四)说明书内容改变。
(五)包装和标签式样、内容改变。
(六)处方中辅料改变。
(七)产地改换。
(八)药品规格改变或增加。
(九)包装规格改变或增加。
(十)其它与批准注册时申报内容有任何改变的。
第二十八条 补充申请需填写《进口药品补充申请表》,连同《进口药品申报资料细则》规定的补充申请资料,报国家药品监督管理局审查批准。
第二十九条 申请增加适应症,须在中国进行临床试验;药品质量标准改变的,须进行质量标准复核。
第三十条 改换产地、增加药品规格的补充申请,必须在原《进口药品注册证》有效期满至少12个月前提交;不足12个月的,可按照本办法第四章的规定,申请换发《进口药品注册证》,同时申请改换产地、增加药品规格。
改换产地、增加药品规格的补充申请经国家药品监督管理局审查批准后,核发新产地、新增药品规格的《进口药品注册证》,新注册证号为原注册证号前加字母B构成,注册证有效期以原注册证为准。
第三十一条 《进口药品注册证》规定内容的补充申请,如变更包装规格、通用名称、商品名、公司名称、生产厂名称等,国家药品监督管理局审查批准后,核发新的《进口药品注册证》,原注册证即行作废,并由国家药品监督管理局收回;增加包装规格的补充申请,包括进口分装生产所需大包装规格,国家药品监督管理局审查批准后核发新的《进口药品注册证》,原注册证可继续使用。
新注册证号为原注册证号前加字母B构成,注册证有效期以原注册证为准。
第三十二条 进口药品中文说明书和质量标准的补充申请,经国家药品监督管理局审查批准后,下发修订后的说明书和质量标准,原说明书和质量标准即行废止。
第三十三条 进口药品的包装、标签的式样和内容仅有微小改变的,应向国家药品监督管理局申报备案。
第六章 药品名称、包装、标签和说明书
第三十四条 进口药品必须使用中文药品名称,必须符合中国药品命名原则的规定。
第三十五条 进口药品的包装和标签,必须用中文注明药品名称、主要成分以及注册证号,进口药品必须使用中文说明书。
第三十六条 进口药品的包装、标签和说明书,必须符合中国《药品包装、标签和说明书管理规定》,并经国家药品监督管理局批准后使用。一经批准,其内容不得擅自更改。
第七章 进口检验
第三十七条 国家药品监督管理局根据进口药品管理工作的需要,设立口岸药品检验所,负责已注册品种的口岸检验工作。
第三十八条 中国药品生物制品检定所负责对进口药品检验工作进行组织、协调和指导,对有争议的检验结果进行技术仲裁,其对进口药品的技术仲裁结果为最终结论。
第三十九条 进口药品必须按照《进口药品注册证》载明的质量标准,逐批全项检验。进口药品检验所需标准品、对照品,由中国药品生物制品检定所负责统一制备、标定和分发。《进口药品检验报告书》实行统一格式。
第四十条 进口药品必须从口岸药品检验所所在城市的口岸组织进口。从其它口岸进口的,各口岸药品检验所不得受理检验。
第四十一条 生物制品的进口检验,由中国药品生物制品检定所负责或国家药品监督管理局授权的口岸药品检验所负责。
第四十二条 进口药品到达口岸后,进口单位须填写《进口药品报验单》(附件三),持《进口药品注册证》(正本或副本)原件,到所在口岸药品检验所报验,并报送以下资料:
(一)加盖进口单位公章的申报品种《进口药品注册证》复印件。
(二)加盖进口单位公章的《药品经营企业许可证》复印件。
(三)申报品种《进口药品注册证》载明的生产国有关部门出具的产地证明原件。
(四)申报品种的购货合同副本。
(五)申报品种的装箱单、运单和货运发票。
(六)申报品种的出厂检验报告书。
(七)申报品种的中、英文说明书,包装、标签和样品。
(八)其他有关资料。
预防性生物制品、血液制品,须同时出具国家药品监督管理局核发的《生物制品进口批件》;进口药材应同时出具国家药品监督管理局核发的《进口药材批件》。
第四十三条 口岸药品检验所在收到《进口药品报验单》后,应及时查验进口单位报送的全部资料,核对药品数量,符合要求的,发给《进口药品报验证明》(附件四)。
第四十四条 海关放行后7日内,进口单位应将已交迄的海关税单报所在口岸药品检验所,并联系到存货地点现场抽样。口岸药品检验所应将所有进口货物数量与海关税单核对一致并完成抽样后,签署《进口药品抽样记录单》(附件五),并将全部货物予以加封。未经检验合格的进口药品不得擅自拆封、调拨和使用。
进口药品抽样,按照国家药品监督管理局《进口药品抽样规定》(附件六)和《进口药材抽样规定》(附件七)执行。
第四十五条 口岸药品检验所抽样后,应及时检验,并在规定时间内出具《进口药品检验报告书》。《进口药品检验报告书》应明确标有“符合规定,准予进口”或“不符合规定,不准进口”的检验结论。需索赔的,应及时出具英文《进口药品检验报告书》。进口检验的样品留存二年。
第四十六条 对检验符合规定的进口药品,口岸药品检验所应及时启封,允许调拨、销售和使用;不符合规定的进口药品就地封存。
第四十七条 进口单位对检验结果有异议时,应在收到《进口药品检验报告书》三十日内向原口岸药品检验所申请复验;如对复验结果仍有异议的,可在收到复验结果三十日内向中国药品生物制品检定所申请仲裁检验。
未申请复验或经仲裁仍不合格的,不合格药品由存货地省级药品监督管理局监督处理。
第四十八条 对检出的不合格进口药品,各口岸药品检验所须在7日内将检验报告书报国家药品监督管理局和中国药品生物制品检定所,同时送其他口岸药品检验所和存货地省级药品监督管理局。中国药品生物制品检定所应在每月第一周将上月不合格进口药品情况统计汇总后报国家药品监督管理局。
第四十九条 下列情形之一的进口药品,其报验申请将不予受理:
(一)不能提供《进口药品注册证》(正本或副本)、及《生物制品进口批件》或《进口药材批件》原件的;
(二)《进口药品注册证》超过有效期30日的;
(三)未提供本次申报品种产地证明原件的;
(四)从事进口业务的单位未取得《药品经营企业许可证》的;
(五)申报品种的包装、标签等与《进口药品注册证》不一致的;
(六)无中文说明书或中文说明书与批准的说明书不一致的;
(七)未在规定口岸进口的;
(八)报验时,药品制剂距失效期不满六个月,原料药、辅料距失效期不满十二个月的;
(九)伪造、涂改有关文件和票据的;
(十)其他不符合进口药品管理有关规定的。
第八章 监督和处罚
第五十条 进口药品必须在《进口药品注册证》载明的生产厂或包装厂完成生产和最终包装。不得在其它国家(或地区)进行生产、改换包装、加贴中文标签和补装中文说明书等。违反上述规定的,口岸药品检验所不予受理其已到岸药品的进口报验。
第五十一条 国内药品生产企业、经营企业以及医疗单位采购进口药品时,供货单位必须提供《进口药品注册证》和《进口药品检验报告书》的复印件,并加盖供货单位的公章。
第五十二条 获得《进口药品注册证》的国外制药厂商,必须按照国家药品监督管理局药品不良反应监察管理的有关规定,及时报告进口药品使用中发生的不良反应,包括在国外发生的不良反应。未按规定报告进口药品的不良反应而造成的一切后果,由国外制药厂商负责。
第五十三条 有下列情形之一的进口药品,禁止销售、使用:
(一)未取得《进口药品注册证》、《生物制品进口批件》或《进口药材批件》进口的;
(二)伪造、假冒《进口药品注册证》、《生物制品进口批件》或《药材进口批件》进口的;
(三)伪造、假冒《进口药品检验报告书》销售的。
第五十四条 有下列情形之一的进口药品,由国家药品监督管理局对国外制药厂商提出警告:
(一)进口检验一批不合格的;
(二)未及时报告药品不良反应情况的;
(三)擅自更改包装和标签的;
(四)包装、标签未注明《进口药品注册证》证号以及未用中文注明药品名称和主要成分的。
第五十五条 有下列情形之一的进口药品,由国家药品监督管理局停止其进口检验:
(一)进口检验二批以上不合格的;
(二)未及时报告药品不良反应,造成严重后果的;
(三)擅自更改国家药品监督管理局批准的中文说明书内容的;
(四)被国家药品监督管理局警告二次以上的(含二次);
(五)超出《进口药品注册证》限定的使用范围的;
(六)已被国外药品主管当局停止生产、销售和使用的;
(七)其它严重违反中国药品管理的法律、法规和规定的。
第九章 附 则
第五十六条 本办法所称进口药品,除原料药、制剂外,还包括制剂半成品和药用辅料等。
第五十七条 申报单位对我局进口药品注册审批的结论有异议的,可在一月内,逞向国家药品监督管理局申请复审。
第五十八条 品、精神药品和放射品的进口管理,按照《品管理办法》、《精神药品管理办法》和《放射品管理办法》的规定执行。
第五十九条 港、澳、台地区生产的药品申请向内地销售、使用的,参照本办法的规定,经国家药品监督管理局批准后,核发《医药产品注册证》。
第六十条 申请进口药品注册须按照国家有关规定缴费。
第六十一条 本办法由国家药品监督管理局负责解释。
第六十二条 本办法自1999年5月1日起实施。有关进口药品的管理规定一律以本办法为准。
附件一:
进口药品申报资料细则
一、申报资料必须符合所附《申请进口药品注册申报资料》、《申请换发进口药品注册证申报资料》和《进口药品补充申请申报资料》规定的资料项目和编号。
二、《进口药品注册申请表》、《换发进口药品注册证申请表》需填写一式三份,《进口药品补充申请表》需填写一式二份,除签字和盖章外,所有内容必须打印。
三、申报资料必须使用中文或英文,其它文种的资料可附后参考。申报资料必须科学、真实、全面,凡字迹模糊、潦草、难以辨认的,一律不予受理。各类中文翻译件必须忠实原文,并符合中国的专业规范。
四、申报品种的各类外国政府药品主管当局证明文件、处方、质量标准、药品说明书需全部译成中文,其它各项研究资料须分别提供各项试验的中文摘要。
五、申报资料须按规定的资料编号顺序整理,不同编号的资料单独装订,并在每项资料的封面上注明资料名称,在右上角注明资料编号。同一资料的中文翻译件应排在原文之前。全部资料应使用A4纸。
六、新申请注册品种编号为A0l一A05的资料并《进口药品注册申请表》装订成一册,为“第一卷”,申请表排在其它资料之前; A06一Al3项资料装订成一册为“第二卷”; A14一A22项资料为“第三卷”; A23一A26项资料为“第四卷”。换发《进口药品注册证》申请以及补充申请的资料,按资料顺序,各装订为一卷,申请表置于其它资料之前。
申报资料每一卷的封面上应用中文注明申报品种的通用名称和商品名、生产公司、申请公司、申报日期以及该卷分属的卷号。各卷首页应备有本卷资料的中文目录。
七、A01项资料必须报送原件,应符合世界卫生组织(WHO)推荐的统一格式。必要时,所报文件需经所在国公证机关和驻所在国中国使、领馆公证。
1.区域国际组织批准药品注册、生产、销售及符合药品生产质量管理规范(GMP)的证明文件,经国家药品监督管理局特别承认,可作为一国的证明文件使用。
2.申报品种在一地完成制剂生产,转由另一地完成包装的,须提供包装厂所在国药品主管当局签署的关于该包装厂符合GMP的证明文件。
3.制剂类申报品种在生产国尚未获得注册批准和销售许可的,可提供其公司总部所在国和其它至少一个国家出具的证明文件,但GMP证明文件必须由生产厂所在国药品主管当局签署。
4.原料药和辅料,按照生产国对此类产品的管理要求,无法提供有关证明文件的,可提供生产国或出口国使用该原料药、辅料制成制剂的证明文件,或提供本国其它有关政府部门出具的证明该品确已上市销售供药用的证明文件。
5.申请进口注册的原料药或制剂半成品为专供中国国内药品生产企业用于生产一类新药,且该企业已获得国内药品生产批准文号,其注册和销售的政府证明文件可报送我国批准该药的新药证书和新药批件的复印件,其GMP证明文件和出口证明必须由国外生产厂所在国药品主管当局签署。
6.部分非处方药(OTC)和医疗营养药品无法提供国家级证明文件的,可按生产国和出口国的法律规定,提供相应级别政府部门的证明文件。
八、国外制药厂商在中国尚未设立办事机构的,必须选定中国代理商代理申报,其A02、H02项资料应报送国外制药厂商授权中国代理商代理申报的授权文书、公证文件及其中文译本,并附中国代理商的工商执照复印件。
如国外制药厂商已在中国设立办事机构,则该办事机构可直接申报,其A02、H02项资料应报送中国工商管理部门核发的该办事机构《外国企业常驻中国代表机构登记证》复印件。
九、首次申报的原料药或制剂半成品,必须报送全部技术资料,其中A23、 A24项资料可报送用本原料药或制剂半成品制成制剂的试验资料。
已有同类品种进口注册的原料药或制剂半成品, A14一A22项资料可报送文献资料, A23、A24项资料必须报送用本原料药或制剂半成品制成制剂的试验资料,不得仅使用文献资料。
十、首次申报的药品制剂,必须报送规定的全部技术资料,不得仅使用文献资料。
已有同类品种进口注册的药品制剂, A14一A22项资料可报送文献资料, A23、A24项资料必须报送所申请品种的试验资料,不得仅使用文献资料。
十一、申请注册药用辅料,除单独使用辅料无法进行的试验项目外,应参照本细则的资料要求报送资料。
十二、传统药物和天然药物的A06、H03项资料应详细注明处方中所用每一原植(动)物或矿物的来源,分属的科、属、种和拉丁名、英文名。
十三、申报进口注册的原料药和辅料, A07、H04项资料必须详细说明其生产工艺和生产流程。
十四、生物制品的A09、 H06项资料应包括生产检定用菌毒种、细胞株以及生产检定用动物、主要原材料等技术资料。
十五、Al2、 H09项资料为拥有申报品种上市权公司质量控制部门的实验室所作检验的报告,由质控部门的负责人签字生效。
十六、A13项资料为申报注册品种三批样品室温留样的稳定性试验资料和加速试验的稳定性试验资料。必须包括全部试验数据,如质量检验报告书和定性、定量色谱分析图谱等。
十七、疫苗制品的Al4项资料包括免疫学资料。
十八、 A23项资料,包括临床试验及人体生物利用度/生物等效性试验资料,其中临床资料应为I期、II期和III期试验资料,包括临床试验方案和统计方法;IV期临床试验资料可申报作为参考。
十九、A24、H10项资料必须提供生产国药品主管当局批准的英文说明书并附中文译本。中文说明书必须严格按照英文说明书的内容翻译,并符合中国的专业规范。如英文不是生产国官方文字,无法提供英文资料的,则必须提供生产国药品主管当局批准的原文说明书,同时须提供其英文译本和中文说明书。
二十、A25、H13项资料为该药品在国外市场上所使用的包装、标签和说明书实样,不包括药品本身; A26、H14项资料为国外市场上所销售药品的实样,包括包装、标签、药品本身和包装盒内的说明书。
二十一、补充申请中有关政府证明文件和技术资料的要求,按照上述规定执行。
申请进口药品注册申报资料项目
A0l 药品生产国国家药品主管当局批准药品注册、生产、销售、出口及其生产厂符合药品生产质量管理规范(GMP)的证明文件、公证文件及其中文译本
A02 国外制药厂商授权中国代理商代理申报的授权文书、公证文件及其中文译本,中国代理商的工商执照复印件;国外制药厂商常驻中国代表机构登记证复印件
A03 药品专利证明文件
A04 该药品在国外的生产、销售和使用情况,生产厂基本概况以及申请在中国注册需特别说明的理由及其中文译本
A05 药品各项研究结果的综述及其中文译本
A06 药品成分和/或处方组成及其中文译本
A07 药品生产工艺及其中文译本
A08 药品质量标准和检验方法及其中文译本
A09 制剂用原料药的质量标准和检验方法及其中文译本
A10 所用辅料的质量标准和检验方法及其中文译本
A11 直接接触药品的包装材料的质量标准和检验方法及其中文译本
A12 三批药品的质量检验报告书
A13 稳定性试验资料
Al4 主要药效学试验资料
A15 一般药理研究资料
Al6 急性毒性试验资料
Al7 长期毒性试验资料
Al8 致突变试验资料
A19 生殖毒性试验资料
A20 致癌试验资料
A21 依赖性试验资料
A22 动物药代动力学资料
A23 药品临床研究资料
A24 药品生产国国家药品主管当局批准的英文说明书(或原文说明书)及其中文译本
A25 药品包装、标签和说明书实样
A26 药品实样和其它资料
申请换发《进口药品注册证》申报资料项目
H01 药品生产国国家药品主管当局批准药品注册、生产、销售、出口及其生产厂符合药品生产质量管理规范(GMP)的证明文件、公证文件及其中文译本
H02 国外制药厂商授权中国代理商代理申报的授权文书、公证文件及其中文译本,中国代理商的工商执照复印件;国外制药厂商常驻中国代表机构登记证复印件
H03 药品现行处方组成及其中文译本
H04药品现行生产工艺及其中文译本
H05 药品现行质量标准和检验方法及其中文译本
H06 制剂用原料药现行的质量标准和检验方法及其中文译本
H07 所用辅料的现行质量标准和检验方法及其中文译本
H08 直接接触药品的现行包装材料的质量标准和检验方法及其中文译本
H09 近期三批药品的质量检验报告书
Hl0 药品生产国国家药品主管当局批准的现行英文说明书(或原文说明书)及其中文译本
土耳其原料药出口注册流程
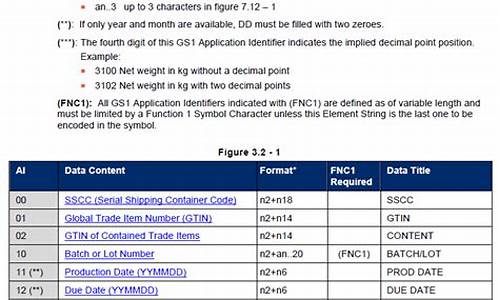
一、在高效液相色谱法系统适用性试验中,有常用的四个内容:分离度、柱效、重复性和拖尾因子。
1、分离度(resolution,R)——相邻两峰的保留时间之差与平均峰宽的比值。也叫分辨率,表示相邻两峰的分离程度。R≥1.5称为完全分离。
2、柱效:色谱柱是色谱仪最重要的部件(心脏)。柱子内径一般为1~6 mm。常用的标准柱型是内径为4.6 或3.9mm ,长度为15 ~30cm 的直形不锈钢柱。填料颗粒度5 ~10μm ,柱效以理论塔板数计大约 7000 ~10000。减小填料粒度和柱径以提高柱效。
3、拖尾因子(tailing factor,T),用以衡量色谱峰的对称性。也称为对称因子或不对称因子。
二、目的:按各品种项下要求对仪器进行适用性试验,即用规定的对照品对仪器进行试验和调整,应达到规定的要求;或规定分析状态下色谱柱的最小理论板数、分离度和拖尾因子。
扩展资料
高效液相色谱法(High Performance Liquid Chromatography \ HPLC)又称“高压液相色谱”、“高速液相色谱”、“高分离度液相色谱”、“近代柱色谱”等。
LLPC与GPC有相似之处,即分离的顺序取决于K,K大的组分保留值大;但也有不同之处,GPC中,流动相对K影响不大,LLPC流动相对K影响较大。
百度百科-高效液相色谱法
美国药物主文件档案 表示什么?
土耳其原料药出口注册流程
原料药是我国医药产业参与国际竞争的优势长板,新冠疫情发生以来,全球医药产业链受到冲击,使得我国原料药产业的战略地位进一步提升。据国家发改委统计,我国几乎可以生产全部品种的原料药,种类高达1500多种,其中60%产量用于出口。截止到2019年,我国原料药共出口到189个国家和地区,主要集中于亚洲、欧洲和北美洲三大市场,国际认证是出口必不可少的一道手续。原料药出口需要递交大量的产品相关资料给当地政府或相关机构获得认证后才能出口,如果不是相同语言的国家出口原料药,所递交的资料和各类说明文件还要进行翻译。原料药认证所需的相关资料对格式要求非常严格,并且专业术语多、难度大。
原料药认证翻译拥有以下资质能更快更好地获得认证:
翻译人员是药学或医学相关专业
熟悉各国的认证流程和相关政策
熟悉认证资料格式和相关术语
国际上常见的三种原料药认证分别为美国的FDA认证、欧洲EDQM认证和日本PMDA认证
FDA认证
美国FDA认证分为五类:I型-生产地点和厂房设施、II型-中间体、原料药和药品、III-型包装物料、IV-型辅料、着色剂、香料、香精及其他添加剂、V-型非临床数据资料和临床数据资料
一般情况下中国厂商的申报认证程序如下:
显示所有大图
EDQA认证
欧洲的CEP 只适用于已有《欧洲药典》( EP) 标准的原料,即 EP 收载的原料药品种。
申请CEP的基本程序包括:
PMDA认证
日本的PMDA认证只能通过日本国内的管理人将MF申请递交给PMDA官方,不能由自己直接递交。
申请MF的基本程序包括:
无论是哪一个国家的申请都离不开CTD文件、GMP检验报告、官方提出的修改意见及问题和补充文件及回复说明。各国文化背景的不同使语言成为了横亘在中间最急需攻克的难题。
国家
修改意见及问题
补充文件及回复说明
美国
483表问题
现场检查及483表的回复说明
欧洲
EDQM提出的修改意见
GMP检查的回复补充说明
日本
PMDA提出的缺陷问题
对PMDA提出的缺陷进行回复
CTD的排版要求
CTD中信息的表达要明确、清楚,申请人不应修改CTD的整体结构,以利于审查内容和快速查找。
纸张大小:欧洲和日本-A4,美国letter纸(8.5X11``)文档和表格应留出余地,以方便在纸张上打印。左手边空白部分应保证装订不受影响
字体:文档和表格的字符大小应足以清楚阅读,建议描述性文档采用Time New Roman,12的字符。
缩写词应在每模块中第一次使用时进行定义。
每页必须有编号页码。
美国DMF文件M1要求
CUVER LETTER(首页)
STATAMENT OF COMMITMENT(声明信)
Administrative Page(行政信息)
US Aent Appointment Letter(美国代理人的指定)
Letter of Authorization(授权信)
Holder Name Transfer Letter(证书持有人转移)
New Holder Acceptance Leeter(新持有人接受函)
REQUEST TO(WITHDRAW,CLOSE) a DMF(DMF的取消与关闭)
Paten satement(专利声明)
欧洲CEP申请文件M1要求
申请表
letter ofAuthorisation(授权信)
declaration in cases where the manufacturer is not the intended holder of a Certificate of Suitability(证书持有人与生产厂商不同的声明信)
letter of declaration of willingness to be inspected(愿意接受检查声明)
letter of declaration of substances of animal/human origin(TSE风险的声明)
letter of commitment to provide samples upon requert by the EDQM(愿意提供样品的声明
模块M2:质量综述
项目
2.3.S.1基本信息
2.3.S.1.1药品名称
2.3.S.1.2结构
2.3.S.1.3理化物质
2.3.S.2生产信息
2.3.S.2.1生产商
2.3.S.2.2生产工艺和过程制作
2.3.S.2.3物料控制
2.3.S.2.4关键步骤和中间体控制
2.3.S.2.5工艺验证和评价
2.3.S.2.6生产工艺的开发
2.3.S.3结构确证
2.3.S.3.1结构和理化性质
2.3.S.3.2杂质
2.3.S.4原料药的控制
2.3.S.4.1质量标准
2.3.S.4.2分析方法
2.3.S.4.3分析方法的验证
2.3.S.4.4批检验报告
2.3.S.4.5质量标准制定依据
2.3.S.5对照品
2.3.S.6包装材料和容器
2.3.S.7稳定性
2.3.S.7.1稳定性总结
2.3.S.7.2上市稳定性承诺和稳定性方案
2.3.S.7.3稳定性数据总结
模块M3: 质量部分
项目
3.1目录
3.2.S原料药
3.2.S.1一般信息
3.2.S.1.1命名
3.2.S.1.2化学结构
3.2.S.1.3一般特性
3.2.S.2生产
3.2.S.2.1生产商
3.2.S.2.2生产工艺和过程控制的描述
3.2.S.2.3物料控制
3.2.S.2.4关键工艺步骤和中间体的控制
3.2.S.2.5工艺验证
3.2.S.2.6生产工艺的改进与变更控制
3.2.S.3结构表征
3.2.S.3.1结构表征和其他特性
3.2.S.3.2杂质
3.2.S.4原料药控制
3.2.S.4.1质量标准
3.2.S.4.2分析方法
3.2.S.4.3分析方法验证
3.2.S.4.4批分析报告
3.2.S.4.5质量标准合理性分析
3.2.S.5对照品
3.2.S.6包装容器和密封方式
3.2.S.7稳定性实验
3.2.S.7.1稳定性实验概述和结论
3.2.S.7.2申请批准后的稳定性实验方案和稳定性保证
3.2.S.7.3稳定性实验结果列表
雅瑞思医学翻译一直专注于医药、医疗器械领域翻译,拥有上百名药学、医学、物理学、生物学、制造学、工程学专职翻译专家。医药翻译部由药理学、毒理学、生药学、药物化学、药物分析学、药剂学、临床医学、临床药理学背景的专业翻译人员组成,都曾在药厂或者大学药学院工作、学习过,对原料药DMF、IND、AND的全套文件翻译极为熟悉,多数成员都具备医学或者药学硕士及以上文凭。
处方上的用量翻译。清楚点哦。我背背,谢谢。
DMF文件简介
美国药物主文件档案(Drug Master File)简称DMF文件,是根据美国联邦管理法的
规定,对于化学原料药中间体、赋料、医药包材等非直接药品进入美国时,须向美
国FDA申请注册并递交有关文件。该文件是由生产商提供的某药品生产全过程的详
细资料,以便于FDA对该厂产品加以全面了解,文件内容包括:生产、加工、包装
和贮存某一药物时所用的具体厂房设施和监控的资料。企业上报的DMF文件原件在
FDA收到后经初审,如符合有关规定的基本要求,FDA就会发通知函并颁发给一个D
MF登记号。
DMF文件共有五种类型:Ⅰ型,生产地点和厂房设施、人员(现已不使用);
Ⅱ型,中间体、原料药;Ⅲ型,包装材料;Ⅳ型,辅料、赋形剂、着色剂、香料、
香精及其他添加剂;Ⅴ型,非临床数据资料和临床数据资料。
国内原料药生产企业向FDA申报的DMF文件属于Ⅱ型,申请文件的主要内容有:
递交申请书、相关行政管理信息、企业的承诺声明、申请产品的物理和化学性质描
述、产品生产方法详述、产品质量控制与生产过程控制、产品稳定性实验、包装和
标签、标准操作规程、原材料及成品的贮存与管理、文件管理、验证、批号管理制
度、退货及处理。
载说明:刚接触医药方面的说明书时,如见天书。好歹也是学习英语超过10年,作为非英语专业、该过的级也过了。大学也是以重视英文教学著称;心理所受打击可想而知。
借着手头上的医学英汉词典,才能看懂。也明白医学和药学其实是很不同的两块领域。
以下的英文药品说书的写法,是我手头上能找到的最好的版本。不仅帮助我了解药品说明书、并学着做说明书的中译英。还帮助理解其它专业英语。]
英文药品说明书的写法——第一节 药品名称
一、进口药英文说明书的结构简介
“药品说明椤钡挠⑽谋泶锓绞接蠭nstructons,Directions,Description 现在多用Package Insert,或简称 Insert,也有用Leeflet或Data Sheets。Insert原意为“插入物,插页”。药品说明书即为附在每种药品包装盒中的一份用药说明。经过注册的进口药品一般是国家承认的有效药物,其说明书是指导医生与患者合理用药的重要依据,具有一定的法律效力。
进口药的英文说明书随药品来源的不同,有以英语为母语的国家,也有以英语为外语的国家。说明书繁简难易不同。短者仅百余词,长者可达上万词。较简单的悦明书仅介绍成分、适应症、禁忌症、用法与用量等内容;较详尽的说明书中除上述内容外还包括:药品性状、药理作用、临床药理、临床前动物试验、临床经验、药代动力学、庄意事项、不良反应或副作用、用药过量、药物的相互作用、警告、有效期、包装、贮存条件、患者须知及参考文献等诸多项目。
为了顺利阅读和正确翻译进口药英文说明书,读者除应具备较好的英语基础,掌握一定的专业知识(如医学、化学、药剂学、药理学、药物代谢动力学等)外,还应熟悉英文药品说明书的结构及语言待点等。大多数英文说明书都包括以下内容;①药品名称(Drug NameS),②性状(Description),③药理作用(Pharmacological Actions),④适应症(Indications),⑤禁忌证(Contraindications),⑥用量与用法(DOsage and Administration).⑦不良反应(Adverse Reactions)。⑧注意事项(Precautions),⑨包装(Package),⑩贮存(Storage),⑾其他项目(Others)。
现将各项专题的表述方法与翻译、结构特点、常用词语及阅读技巧等分述如下。
二、药品名称(第一节)
英文药品说明书中常见的药品名称有商品名( Trade Name或 Proprietary Name),通用名( Generic
Name)和化学名(Chemical Name),其中最常见的是商品名。例如,日本田边有限公司生产的熊去氧胆酸
片,其商品名为 Ursosan(Tablets):通用名为 Ursodesoxycholic Acid(熊去氧胆酸);化学名为3a,7p dihydroxy-5p-Cholanoic acid(3a,7p二羟基5p胆烷酸)。有时同一种药品,不同的厂家使用不同的商品名称。
药品说明书中的标题药名用其商品名。有时在其右上角(或在上角)有一(R)标记,例如ADRIBLASTNA(R)(阿霉素),TEGRETOL(痛痉宁)。“R”是Register(注册)的缩写,(R)表示该产品已经本国的有关部门核准.取得了此项专用的注册商标(Registered Trade Mark)。有时在商品名之下(或后)又列有通用名或化学名.例如: Rulide(罗力得)之下列有(Roxithromycin,罗红霉素): Minipress(脉宁平)之后列有(Praxosin HCI,盐酸哌唑嗪); Nitro-Dur(护心贴片〕之下又列有( Nitroglycerin,)。
药品名称的翻译可采用音译、意译、音意合译及谐音译意等方法。
1、音译:按英文药品名歌的读音译成相同或相近的汉语。如:Tamoxitn它莫西芬,Ritalin利他林,Am-
cacin 阿米卡星。音译较为方便,但不能表意。
2、意译:按药品名称所表达的含意译成相应的汉语。例如:cholic Acid 胆酸,Tetracyline四环素;也可
按其药理作用翻译.如:Minidiab灭糖尿(治疗糖尿病药物),Natulan疗治癌(细胞生长抑制剂),Uraly消石素(治疗尿路结石药物)等。
3、音意合译:药品名称中的一部分采用音译,另一部分采用意译.例如:Coumadin香豆定(coumarin香
豆素),Neo-Octin新握克丁(neo-新);Medemycin麦迪霉素(-mycin 霉素),Cathinone卡西酮(-one酮)。
4、谐音译意:以音译为原则,选用谐音的汉字,既表音,又表意,音意结合。例如:Antrenyl安胄灵,Doriden多睡丹,Legalon利肝隆,Webilin胃必灵.商品名称可以这样翻洋,而法定名称则规定不可以这样翻译。
药品的化学名称反映出该药品的化学结构组成成分,可借助英汉化学化工词典进行翻译。如果名称很长,可以分解开来,分别查出各个组成部分的名称,组合而成。例如:Catalin(卡他林)的化学名称是1-Hvdroxy- 5-oxo-5H-pyrido(3,2-a)-Phenoxazine-3-carboxylic acid,译成汉语是1-羟基-5-氧-5H-吡啶开(3,2-a)吩 嗪-3-羧酸。如能掌握一些常用的酸、碱、盐、基因、化合物的英文名称,以及常用的前缀、后缀等,翻译时会顺利得多。例如:chloride氯化物,sulfate(sulphate)硫酸盐,acetyl一乙酰基,amino氨基,di-二,dihydro-二氢。nitro-硝基,-ester酯,-lactone内酯,-one酮、-oxide氧化物,-urea脲等等.
为了统一药品名称的译名,卫生部药典委员会已拟定出原料药和辅料命名原则,并刊行了<药名词汇>一书,可供翻译英文药品名称时参考。
第二节 性状
许多药品说明书的第一项是Description(性状),其原意是“叙述”、“描写”,在药品说明书及药典中一般都译为“性状”,其内容主要是介绍外观、理化性质、组成成分、结构、特征等。这一项最常用的标题是Description,此外还可能有其他的表示法,如:
Chemical Structure 化学结构
Composition 成分
Physical and Chemical Properties 理化性质
这一项中的英语词汇除一部分化学专业词汇外,多为常用词,借助英汉化学化工词典及英汉词典即可通读。
一、本项中常见的句型
例1.Folic acid is a yellowish to orange, crystalline powder; odourless or almost odourless.
叶酸是淡**至橙色结晶粉沫,无臭或几乎无臭。
例2.Intralipos 10% is a white opaque fat emulsion for intravenous injection, containing 10 W/V % of purified soybean oil.
脂肪乳剂(10%)是白色,不透明,供静脉注射用的脂肪乳剂,含有10%(W/V)的精制大豆油。
例3.Ursosan Tablet 50mg is a white plain tablet which contains 50mg of ursodesoxycholic acid.
熊去氧胆酸片为白色素片,每片含50mg熊去氧胆酸。
例4.Sterile pyrogen-free, orange red, freeze-dried powder in vials containing 10mg and 50mg of doxorubicin hydrochloride with lactose.
(本品)为小瓶装,灭菌无热原,桔红色冻干粉沫,每小瓶含有10mg或50mg阿霉素盐酸盐与乳糖。
例5.It occurs as a white to off-white, crystalline solid, poorly soluble in water, dilute acid and most organic solvents.
本品(炎痛息康)为白色至类白色结晶固体,难溶于水、稀酸及大多数有机溶剂中。
例6.Pamine, chemically known as epoxytropine tropate methylbromide, has the empirical formula C18H24NO4Br and the molecular weight 398.3.
哌明的化学名称为环氧莨菪碱托品酸酯溴代甲烷,实验式为C18H24NO4Br,分子量为398.3。
例7.Kanendomycin is a very stable antibiotic, and its activity does not decrease when the powder is placed in an airlight container and kept at room temperatures for more than 2 years.
卡内多霉素是一种很稳定的抗生素,其粉沫置于密封容器中,在室温下保存二年以上,活性不减。
例8.This product is prepared from units of human plasma which have been tested and found nonreactive for hepatitis associated (Australia) antigen.
本品由人血浆制备,此血浆业经检验,并且证明对肝炎(澳大利亚)抗原无反应。
二、本项中常用的词语
1、表示组成、制备的词及短语,如:
be derived from 由……衍生
consist of 由……组成
be obtained 制得
contain 含有
be prepared from 由……制备
have (possess) 有(具有)
2、表示性质的一些词类,如:
colo(u)r 颜色
stable 稳定的
taste 味道
molecular formular 分子式
odo(u)rless 无臭的
molecular weight 分子量
crystalline 结晶的
structure 结构
solubility 溶解度
injection 注射剂
insoluble 不溶的
solution 溶液
odo(u)r 气味
tablets 片剂
colo(u)rless 无色的
derivative 衍生物
tasteless 无味的
liquid 液体
sterile 无菌的
powder 粉沫
soluble 可溶的
solid 固体
还有许多其他词汇,不能一一列举。记住这些常用词对阅读本项内容大有益处。
第三节 药理作用
有些说明书较详细地介绍药品的药理作用(Pharmacological Actions)。其内容主要包括药理作用、临床药理(Clinical Parmacology)、体外试验(in vitro experiments)、药物代谢(Metabolism)、药效(Potency)及毒性(Toxicity)等。这一项常用的标题是:
Pharmacological Action 药理作用
Pharmacological Properties 药理性质
Pharmacology 药理学
Clinical Pharmacology 临床药理
其他的表示方法还有:
Actions 作用
Actions and Properties 作用与性质
Clinical Effect (Use) 临床效果(用途)
Mechanism of Action 作用机理
Mode of Action 作用方式
如果药品的一种抗生素,可能出现:
Biological Action 生物活性
Microbiology 微生物学
此外,还有一此其他的表示方法,这里不一一列举。
这一项目中涉及的词汇范围包括药理学、生理学、化学、毒理学、微生物学及医学等学科,专业词汇多,是较难阅读的一部分内容,阅读时可参阅《英汉医学词汇》、《英汉微生物学词汇》及《英汉化学化工词汇》等工具书。另外,还会遇到许多缩写词,如:CNS(中枢神经系统)、EEG(脑电图)、LD50(半数致剂量)、ECG(心电图)等,这些缩写词可在英汉医学缩略语词典中查到。
一、常见句型举例
例1. Mean peek serum concentrations of tobramycin occur between 30 and about 60 minutes after intramuscular administration.
肌注后约30~50分钟之间妥布毒素的平均血药浓度达到高峰。
例2. Nembutal Sodium exerts a depressant action on the CNS and shares the sedative-hypnotic actions typical of the barbiturates.
戊巴比妥钠对中枢神经系统产生抑制作用,并显示戊巴比妥类特有的镇静催眠作用。
例3. In clinical trials the drug was shown to be highly effectinve in improving and normalizing the alterated cerebral circulation and those disorders related to insufficient arterial flow in the limbs.
临床试验证实,本品疗效高,可改善已改变了的脑循环,使之恢复正常,治疗与四肢动脉血流不畅有关的疾病。
例4. Orbenin is stable to staphylococcal penicillinase, and highly effective against resistant staphylococci.It is bactericidal, acid-stable and well absorbed by either the oral or the intramuscular route.
全霉林对葡萄球菌的青霉素酶稳定,对耐药葡萄球菌十分有效。本品具杀菌、耐酸作用,且口服或肌注吸收良好。
例5. Nystain has been found to inhibit the growth of yeast like flora in the intestinal tract.
已查明制霉菌素在肠道内可抑制菌丛类酵母菌的生长。
例6. Fenarol has proved to be effective as a striated muscle relaxant.
已证明芬那露是疗效很好的横纹肌松施药。
例7. Halcion is a potent short-acting hypnotic agent, which produces its hypnotic activity from the first night of administration.
好而睡是一种强力速效催眠药,它从服药后的第一个夜晚开始产生催眠作用。
例8. Therapeutically, ATP injection exhibits effects, especially such as activation of the function and metabolism of the nerve, and also coronary and peripheral vasodilation to increase the blood stream.
从治疗上看,三磷酸腺苷注射剂显示了非常好的效果,特别是在活化神经的功能及代谢,以及舒张冠状与外周血管以增加血流方面更是如此。
二、常用词及短语举例
1、动词
absorb 吸收
act 作用
cause (be cause by) 引起(由……引起)
demonstrate 显示
exert (action on) 起……作用
exhibit 显示
inhibit 抑制
accumulate 积蓄
administrate 投药
excrete 排泄
result in 导致 indicate 表明
maintain 维持
produce 产生
protect (from) 保护(不变)
reach 达到
show 显示,表明
treat 治疗
metabolize 代谢
promote 促进
prevent 阻止,预防
tolerate 耐受
2、形容词
(be) active (effective) against 对…有效的
(be) related to 与……有关的
(be) sensitive to 对……敏感的
resistant to ……有耐药性的 average 平均的
minimum 最低(小)的
maximum 最高(大)的
normal 正常的
3、名词
ability 能力
activity 活性
distribution 分布
excretion 排泄
action 作用
clearance 廓清率
effect on 对…的作用
function 功能,作用
half life 半衰期
in vitro 体外 kidney 肾
mechanism 机理
serum concentration 血清浓度
tolerance 耐受性
infection 感染
in vivo 体内
level 水平,浓度
plasm lever 血浆浓度(水平)
toxicity 毒性
以上仅举部分例词,此外还有许多专业词汇和基础词汇请参阅有关资料。
声明:本站所有文章资源内容,如无特殊说明或标注,均为采集网络资源。如若本站内容侵犯了原著者的合法权益,可联系本站删除。